
Abstract

The New England Journal of Medicine © 2014
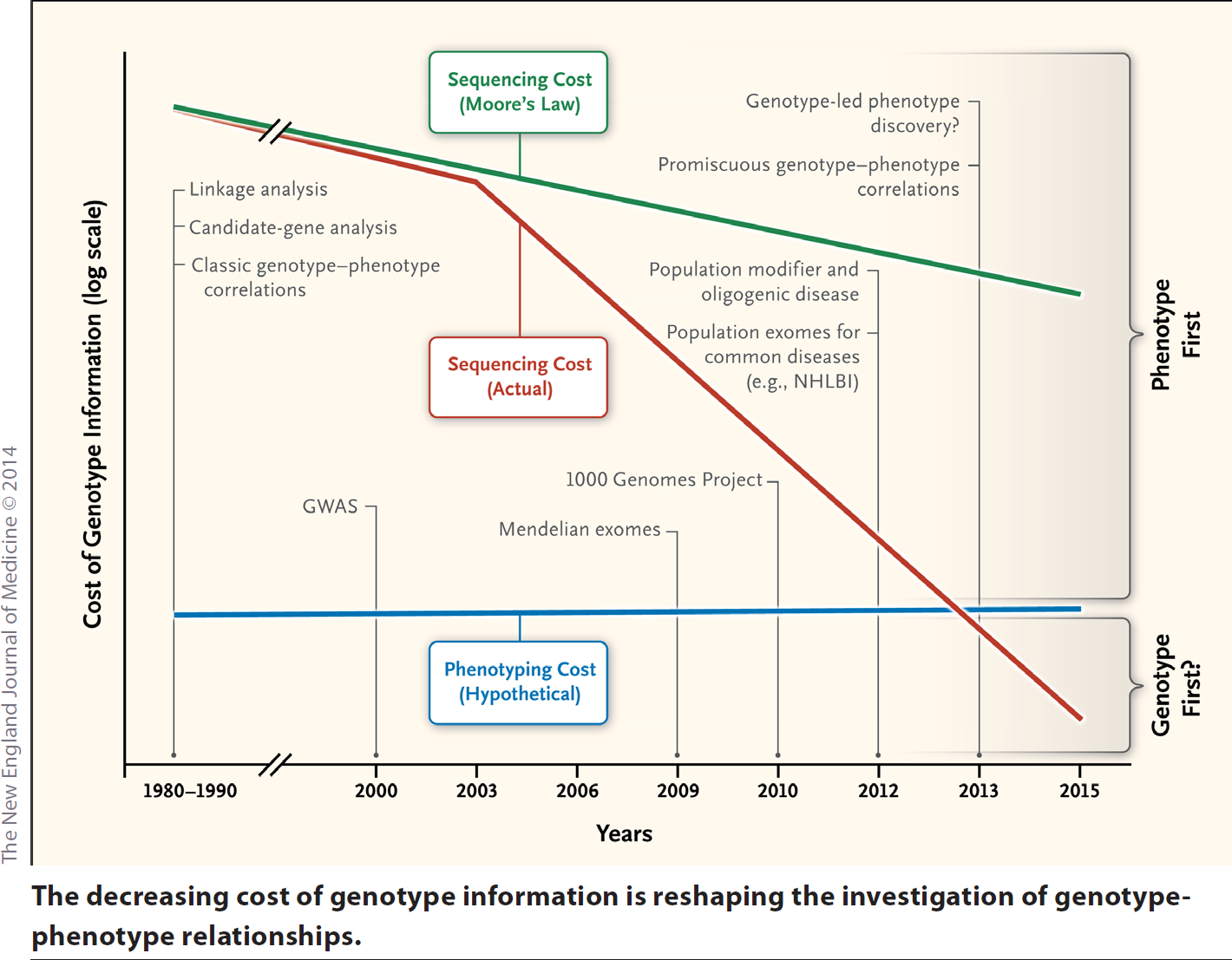
Clinical geneticists from Baylor College of Medicine and the University of Montreal suggest that the “genotype first” era may have finally succeeded the “phenotype first” era of disease discovery—at least so far as Mendelian disease gene identification is concerned. These scientists, in a Perspective article contributed to the New England Journal of Medicine, emphasize that while phenotyping costs have remained relatively flat, sequencing costs have plummeted. In fact, say the authors, sequencing costs are falling even faster than Moore's law might have indicated.
The Perspective article, entitled “Genotype-Phenotype Correlation— Promiscuity in the Era of Next-Generation Sequencing,” presents a graph in which the trace for sequencing costs over time slips below the corresponding trace for phenotyping costs sometime late in 2012. The transition must have occurred suddenly. The previous year, in a review article published by Genome Biology, Dutch researchers weighing the pros and cons of Mendelian disease gene identification approaches, noted that whole-exome sequencing was disadvantaged because it was “relatively expensive.” Whole-genome sequencing, they added, was “even more expensive.”
Just to be clear, the authors of the Perspective—Baylor's Brendan Lee, M.D, Ph.D., and James T. Lu, Ph.D., and Montreal's Philippe M. Campeau, M.D.—have not written a work meant to satisfy accountants or economists. (Their graph's cost axis omits numbers.) Instead, they base their argument on a fairly straightforward observation: next-generation sequencing, especially whole-exome sequencing, has not only become more affordable, it has resulted in an explosion of discoveries of novel genetic mutations.
When the authors refer to the “promiscuity” of genotype-phenotype relationships, they seem to be saying that a particular disease presentation may correspond to myriad genetic mutations or, rather, different collections of subtle genetic changes. Also, a particular genetic mutation, in different contexts, could be associated with different diseases.
In other words, the staid genotypephenotype relationship of old—the unmistakable association between a highly penetrant mutation and an obvious functional change—is just a small part of the emerging disease-gene relationship scene. Even presumably simple Mendelian diseases appear to harbor previously unsuspected first complexities—allelic heterogeneities and locus heterogeneities masked by the overlapping of clinical phenotypes.
“For many Mendelian diseases, single genetic variations (e.g., single-nucleotide polymorphisms, frame-shift insertions and deletions, triplet repeats, and copy number variants) are often good predictors of clinical disease,” wrote the authors. “Yet for most diseases (both common and complex disorders), prediction of clinical and treatment prognoses is challenging because of complex genetic mechanisms and variable expressivity and penetrance.”
One specific example of genotypephenotype promiscuity, said Dr. Lee, concerns brittle bone disease: “We used to think that if you have frequent bone fractures and loose connective tissue, you have brittle bone disease caused by genetic lesions in type I collagen. Now we know that mutations in more than 13 genes can contribute to the cause of brittle bone disease. There can be really rare patients with a mutation in a very specific gene which alters its function in a unique way—not simply loss or gain of the normal function of this gene.”
Such findings encourage researchers to consider more elaborate disease mechanisms. Again, with reference to brittle bone disease, the Perspective explained that “secondary causative and modifier alleles seem to conform to the model of clan genomics or mutational burden.” These alleles “have rare, recent deleterious mutations that, though individually necessary, are not sufficient to cause disease without other mutations.”
While the authors acknowledged the inherent complexity of promiscuous genotype-phenotype correlations, they seemed confident that “continued collection, characterization, and sequencing of Mendelian and common complex diseases will provide new opportunities to unravel the developmental and homeostatic mechanisms governing specific tissues.” The challenge going forward, the authors wrote, lies in “determining what combination of statistical proof with high-and low-throughput in vitro and in vivo validations will be required before combinations of rare variants in multiple genes are accepted as pathogenic.”