
Abstract

Paul Dougall, Ph.D., Oxford Gene Technology

Identifying the causal genetic origins of intellectual disabilities and developmental disorders (ID/DD) is highly complex. One of the reasons for this complexity is the wide variety in the types of genomic aberrations underlying these conditions. Many conditions in this category are, either fully or partially, caused by large aberrations such as copy number variation (CNV) and loss of heterozygosity (LOH). However, small abnormalities such as single nucleotide variants (SNVs) and indels can also play a key role.
De novo mutations are involved in many cases of ID/DD, which are abnormalities that are not inherited from either parent, but occur during or shortly after gametogenesis. The best-known examples of disease-causing de novo mutations are chromosomal aneuploidies such as Down syndrome, but many smaller mutations are also responsible for ID/DD1.
The probability of large de novo CNV causing neurodevelopmental disorders and congenital malformations is often high, in part because one abnormality often disrupts more than one gene. However, the rate at which it occurs in an individual is relatively low, and was recently estimated to arise at a frequency of 0.01 to 0.02 events per generation1.
Very small de novo abnormalities such as SNVs, on the other hand, are much more common. One study estimates the number of de novo SNVs to be around 44 to 82 per individual. Although the effect of one SNV on an individual's development is likely to be low, its high prevalence indicates its importance when looking for underlying causes of ID/DD. Examples of disorders where de novo SNVs play a large role include Rett syndrome (associated with mutations in MECP2) and Alport syndrome (linked to COL4A3 mutations)1.
This large variety in the size of genetic abnormalities highlights the importance for detecting variants of all types and sizes in the field of ID/DD. Array comparative genomic hybridization (aCGH) microarrays are commonly regarded as the gold standard for detecting large abnormalities, typically a few kb and higher. As microarrays cannot detect smaller abnormalities, there is a significant probability that key mutations are overlooked by array-based methods, thus additional testing may be required resulting in a delay to reporting.
To find SNVs and indels efficiently on a large number of loci across the genome, next generation sequencing (NGS) is often used. NGS provides the single nucleotide resolution required for this application and can, depending on requirements, be performed as whole-genome, whole exome or targeted sequencing.
Targeted sequencing is the most efficient way to find underlying causes for ID/DD. It avoids the high time and cost investments associated with whole genome sequencing by studying a large panel of genes associated with ID/DD rather than the entire genome. Using the available sequencing capacity in this targeted manner helps to achieve a high depth of coverage in these selected genes and also helps to avoid finding high numbers of variants of unknown significance (VOUS), which often require extensive additional analysis.
Expanding scope, streamlining workflows
Historically, targeted NGS has not been able to reliably detect large abnormalities such as CNVs—a notion that still persists among many working in the field. However, recent breakthroughs in targeted panel bait design, target enrichment and analysis software have now made accurate and reliable CNV detection possible.
As a result, targeted NGS is now able to offer a fast, comprehensive approach for detecting a broad range of ID/DD abnormalities in one single assay. This approach can save valuable time in finding causative mutations, as well as reducing costs by limiting the need to reflex to other assays.
For sensitive and specific CNV calling NGS panels must have optimal bait design, which can be achieved by spreading baits across the targeted gene regions (at the exon level)—as well as creating a backbone of baits across the rest of the genome to detect larger CNVs and LOH. Small CNV calling is also improved by ensuring the depth of coverage across the targeted genes is as even as possible.
Another pivotal aspect of high-quality results is the software used for analyzing sequencing data. Bioinformatics has long been an integral part of genomic assays, but for many years NGS software has not been optimized for detecting large genetic abnormalities. However, recent improvements in algorithms have led to higher reliability in calling CNVs.
The result of these improvements in NGS methods for ID/DD investigation, is that labs see a substantial increase in the probability of finding a meaningful result in a single assay. The probability is around 15 to 20% for array-based analysis2 (without SNV and indel detection), but could potentially be doubled when using targeted NGS.
Validating outcomes
When considering a change in workflow, such as a move from microarray analysis to NGS, it is imperative to carry out a thorough comparison study to confirm that the new assay performs as well as the existing assay. In the case of studying arrays vs NGS, accurate and reliable CNV detection will be the key indicator.
Figure 1 shows the result of a research sample that had a known deletion analyzed using a targeted NGS panel and a microarray. There was complete concordance between the two methods—highlighting the accuracy of the CNV calling from NGS.
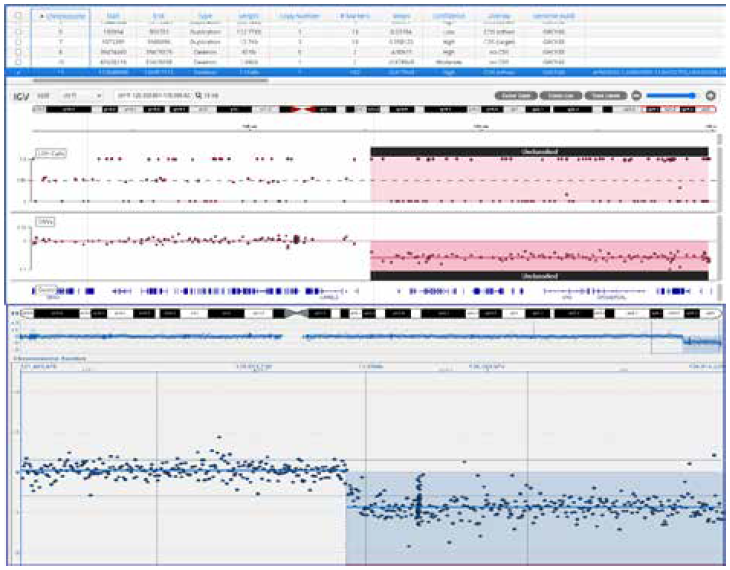
Example of a study comparing microarray analysis and a targeted NGS panel. The region shown in both panels is part of chromosome 11 and includes a 7.3 Mb deletion. Top: CytoSure Constitutional NGS panel. Bottom: CytoSure Constitutional v2 array for developmental disorder research.
Summary
Identifying the genetic origins of ID/DD has evolved significantly over the years—most notably the improvement in resolution when moving from karyotyping and in situ hybridization to microarrays. The probability of finding a cause for ID/DD from a microarray is often around 15 to 20%, which means extra testing and extra spending is necessary to increase the chance of a positive result. Whole genome or exome sequencing is sometimes used as a follow-up test after an inconclusive array result, but is expensive and time-consuming to analyze. Improvements in target enrichment, bait design and software have challenged the perception of NGS being unsuitable as a first-line test.
Constitutional-focused targeted NGS offers a highly suitable, efficient alternative to microarrays in ID/DD applications with simultaneous detection of SNVs, indels, CNV and LOH—while also offering benefits over whole genome sequencing, such as fewer VOUS, lower costs and higher depth of coverage. These capabilities provide confidence when switching from microarrays to targeted NGS, ultimately speeding up workflows and improving the number of actionable results, without compromising on CNV data quality.