
Abstract
Abstract
Realizing that genes often operate together, studies into the molecular biology of cancer shift focus from individual genes to pathways. In order to understand the regulatory mechanisms of a pathway, one must study its genes at all molecular levels. To facilitate such study at the genomic level, we developed exploratory factor analysis for the characterization of the variability of a pathway's copy number data. A latent variable model that describes the call probability data of a pathway is introduced and fitted with an EM algorithm. In two breast cancer data sets, it is shown that the first two latent variables of GO nodes, which inherit a clear interpretation from the call probabilities, are often related to the proportion of aberrations and a contrast of the probabilities of a loss and of a gain. Linking the latent variables to the node's gene expression data suggests that they capture the “global” effect of genomic aberrations on these transcript levels. In all, the proposed method provides an possibly insightful characterization of pathway copy number data, which may be fruitfully exploited to study the interaction between the pathway's DNA copy number aberrations and data from other molecular levels like gene expression.
1. Introduction
CNAs are measured in a high-throughput fashion by array CGH (Pinkel and Albertson, 2005). In an array CGH experiment, differently labeled test (cancer) and reference samples are hybridized together to an array. The reference sample is assumed to have copy number two. Image analysis then results in test and reference intensities. The log2 ratio of the test and reference intensities reflect the relative copy number in the test sample compared to that in the reference sample.
The array CGH data are pre-processed to arrive at an estimate of the copy number of a genomic segment. First, the log2 ratios are normalized (Neuvial et al., 2006). Then, motivated by the underlying discrete DNA copy numbers of test and reference samples, change-point analysis techniques (Olshen et al., 2004) divide the genome into non-overlapping segments that are separated by breakpoints. These breakpoints indicate a change in DNA copy number, and consequently, the copy number does not change within a segment. In addition, the mean log2 ratio of the segments is estimated. Due to the relativity of the measurement, the exact copy number of a segment cannot be determined; however, using mixture model approaches (Van de Wiel et al., 2007) deviations from the normal copy number can be detected. Each segment is then classified as either “normal,” “loss,” or “gain”—“normal” if there are two copies of the chromosomal segment present, “loss” if at least one copy is lost, and “gain” if at least one additional copy is present. These labels are referred to as calls. This classification is not perfect, for example, due to experimental noise or unknown contaminations by normal cells. To address this imperfection, the CNAs are represented by a vector of probabilities, one for each type of call that is discerned. Such probabilities reflect both cell heterogeneity and precision of the array CGH data. We refer to calls and these call probabilities also as “hard” and “soft” calls, respectively.
The potential of call probabilities in the analysis of array CGH data was suggested in Van Wieringen et al. (2007), and demonstrated in Van Wieringen and Van de Wiel (2009) and Gonzalez et al. (2009). Here, the use of call probabilities, through their clear interpretation, enables us to assign meaning (in terms of the copy numbers) to the characterization of a pathway's CNA patterns, which would be difficult when using the log2 ratios. In turn, this meaning is crucial when linking the CNA patterns to, for example, gene expression if it is to provide understanding of this relationship.
We take the call probability signatures at the genomic locations of the genes in a pathway to constitute the copy number data of that pathway. Here a pathway is only a label for what is otherwise known as a gene set. A gene set is a collection of presumably related genes, for instance because they are believed to contribute to the same biological function. Gene set definitions are usually taken from repositories such as GO (Gene Ontology Consortium, 2000) or KEGG (Ogata et al., 1999). It is believed that analysis in terms of functionally related items facilitates the interpretation of results. Note however that such an analysis depends on the quality of the definition of the gene sets, which may have been compiled using incomplete, imperfect or incorrect information (Khatri and Draghici, 2005).
The study of CNAs in terms of pathways demands some justification, for regulatory mechanisms in a pathway are not described in terms of the genomic segments as defined by the breakpoints found in the array CGH experiment. Still, CNAs affect the transcriptome. They exert their influence through the entities (mRNAs, microRNAs, et cetera) that map to the segment. Among others, Pollack et al. (2002) showed the direct (univariate) effect of an increase (or decrease) in copy number on a gene's expression levels. However, genes often operate together (Vogelstein and Kinzler, 2004). Therefore, in order to advance our understanding of a pathway's regulatory mechanisms, one must study its genes at all molecular levels, also at the genomic level. This has been done in, for example, Valentijn et al. (2005) and Ferreira et al. (2008), who both show that copy number changes may affect the pathway's gene expression profile.
This article describes exploratory factor analysis (EFA) designed especially for the call probability array CGH data of a pathway. The EFA characterizes the variability in the CNA patterns by means of a number of latent variables. Hence, EFA is a dimension reduction technique, much like principal component analysis (PCA). PCA has been successfully used to characterize multivariate phenomena in gene expression data, and here we show that EFA has the same potential. Hereto we show that the clear interpretation of the call probabilities is transmitted to the latent variables. This helps to understand what is common and what is not—in terms of their CNA pathway profile—to the samples in the study. Such understanding may also be useful when exploring the effect of copy number changes (as captured by the latent variables) on gene expression levels in the pathway. In two breast cancer data sets, we show how the EFA characterizes copy number data of a GO node, and how it appears to capture the global effect of genomic aberrations on the node's gene expression levels.
1.1. Related work
As far as we are aware no method like exploratory factor analysis tailor-made for array CGH data has been proposed. We acknowledge that PCA could directly be applied to normalized or segmented log2 ratio data, as is done by, for example, Somiari et al. (2004) and Unger et al. (2008). We therefore compare the proposed method to these approaches later and digress here on aspects that set them apart.
The fundamental difference between PCA and factor analysis is that the former does not assume a model for the data, whereas the latter does. This absence (or presence) of a model brings about other differences, for example, in components, eigenvectors (PCA) versus latent variables (EFA), and in the component construction algorithms, singular value decomposition (PCA) versus maximum likelihood type procedures (EFA). Despite their differences, PCA and factor analysis may produce similar results. See Schneeweiss and Mathes (1995) for an account of the mathematical conditions for their results to be similar. Apart from these methodological differences, the assumption of a model also has practical consequences: it facilitates (in combination with the interpretable call probabilities) the interpretation of latent variables. This is of crucial importance if the analysis is to provide insight into the biological phenomenon under study. Note that often (Alter et al., 2000) principal components are assigned interpretations as “eigengenes,” “supergenes,” or “meta-genes.” This interpretation is merely a label, for it is linked neither to a biological entity nor to a theoretical construct. The interpretation of principal components is generally not straightforward, especially if the number of features that contribute to the component gets large.
2. Methods
2.1. Model
Consider an experiment involving a sample of n cancers of a particular tissue. Associated with each cancer sample there is a copy number profile, which we take to consists of the call probabilities. Suppose pre-processing maps the raw array CGH data onto a scale with ordered categories
We also assume the existence of m latent variables. These are introduced to reduce the dimensionality of the copy number data, as it is believed that the information/variability in the full data set can approximated/explained by a much smaller data set. The latent variables are denoted by
We model the conditional expectation of the Qi,j,k given the latent variables as:
In the above model, αj,k is the baseline log probability of call k for region j, and βj,k,ℓ is the change in the kth log probability per unit change in latent variable ℓ for region j. We refer to the βs as the factor loadings. This model is identical to the generalized linear model used for nominal data (McCullagh and Nelder, 2000). Model (1) can also be considered to model the expectation of a Dirichlet distribution with parameters
To ensure all parameters in the model are estimable we set αj,normal = 0 = βj,normal,ℓ for all j and ℓ. For βj,normal,ℓ (similar reasoning applies to αj,normal) the need for this restriction becomes clear when studying the odds of call k1 over call k2, given by:
Thus, the contrast between the vectors
In the above, we have modeled the expectation of the call probabilities directly, and have refrained from specifying their distribution. Two obvious choices, as both are defined on the simplex, are the Dirichlet distribution (Kotz et al., 2000) and the logistic-normal distribution used for compositional data (Aitchison, 1992). Neither provides a reasonable description of the typical patterns of variability of the call probabilities. Figure 1 shows two examples of the distribution of call probabilities.
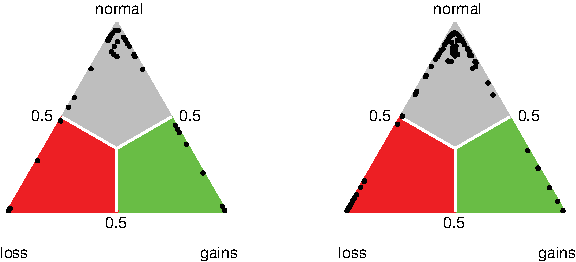
The distribution of call probability data on the simplex of two features.
In call probability data the majority of data points falls on the nodes and edges of the simplex. This is due to the fact that the log2 ratios often clearly indicate (say) a gain. If there is uncertainty in the calling, it is between adjacent calls for example, between normal and gain. A small number of data points falls a little more in the interior of the simplex, corresponding to a probability mass slightly more equally distributed over all calls. This is likely to be due to cell heterogeneity and noisy array elements. Note that the fact that there are no observations near the points
Focusing on a single array element still, the unconditional expectation of Qi,j,k is:
where f the density of the
Assuming the latent variable distribution to be independent standard normal removes the indeterminancy of the model only up to a rotation.
For any rotation matrix
making the corresponding models indistinguishable. This indeterminancy is common to all latent variable models (Bartholomew and Knott, 1999), and is irrelevant for the purpose of low-dimensional plotting, as most statistical software packages allow the user to rotate the axis in order to find an angle of his or her liking. However, the choice of rotation matrix affects the interpretation of the latent variables as brought forth by the factor loadings, but this choice should be made on non-statistical grounds as all rotations fit the data equally well.
2.2. Parameter estimation
The parameters of model (1) are estimated by minimizing the following loss function:
This loss function can be motivated by further pursuing the analogy with nominal data modeling (briefly mentioned in the previous section). Would the calling process have been perfect, the calls are determined without uncertainty, resulting in hard calls (instead of soft calls). In the present notation, the call probability data
We evaluate the integral in (2) by the Gauss-Hermite quadrature approximation:
where the
The parameters are now estimated by minimizing the Gauss-Hermite quadrature approximated loss function. This is done through the application of a modified version of the EM-algorithm proposed in Qu et al. (1996), where it is used to fit a latent variable model to binary data. The EM algorithm as applied to the present situation can be described as follows:
which could (loosely) be interpreted as being (proportional to) the posterior probability of observing call probability profile
with respect to the parameters
The algorithm is initialized with
To assess whether this estimation procedure is capable of reconstructing the latent variables, we conducted a small simulation study (see Supplementary Material). This shows that the Spearman's rank correlation between the “true” factors and their estimated counterparts is close to either one or minus one (the latter is due to the fact that the model is determined up to a rotation).
As with most optimization methods, the above algorithm may converge to local minima. It may therefore be useful to run it several times, the random choice of the initial βs warrants different initial values. It is our experience that the algorithm is quite stable, converging to the same minimum (see Supplementary Material).
2.3. Estimation and interpretation of the factors
Having obtained an estimate of the parameters
which (again) is derived in analogy to the multinomial situation, where it would correspond to posterior expectation.
To assign interpretation to the latent factors we have found the following procedures useful:
Plot the factor scores of the latent variable against a contrast of call probabilities like Select, e.g., four samples, two with the highest and two with the lowest factor score of a latent variable. Compare their call probability profiles. Differences in aberration patterns between the samples with the high and low factor scores may be related to the studied latent variable (see Supplementary Material).
In our experience, based on the analysis of multiple data sets, almost always one factor is highly correlated with the proportion of aberrations (or the like) of the samples. The other factors are more complicated to interpret but seem often to be related to a contrast in aberrations of a particular genomic segment.
The interpretation is however dependent on the rotation chosen. This is irrelevant for the situation with one latent variable. Also for m = 2, as almost always one factor is related to the percentage of aberrations, the rotation is fixed up to symmetry, and consequently the interpretation is fixed. For three (or more) latent variables, one still has a degree of freedom in the rotation. In such cases, to facilitate easy interpretation, the rotation is often chosen to maximize the variance of the (standardized) factor loadings. This results in factor loadings being either close to zero or being away from zero. The latent variable then only has to be interpreted in terms of the features corresponding to the latter as they have the largest contribution to the latent variable. Note however that the larger the number of factors (three and higher), the more difficult to endow all of them with a clear interpretation.
2.4. Ranking
In the event that one studies not a particular pathway but many pathways simultaneously, it is of interest to rank those that are best characterized by the results of the exploratory factor analysis. Hereto we present two statistics both measuring the quality of the EFA characterization that can be used to rank the pathways. The first criterion is the loss reduction (LR) obtained by minimizing (2):
where
which measures the discrepancy between the observed call probability data and a distribution g(Qi,j,k). The cross-entropy between the data and the fitted model (CEfitted) is calculated by replacing g(Qi,j,k) by
3. Illustrations
The potential of the proposed method is illustrated by an exploratory study of two breast cancer data sets published by Pollack et al. (2002) and Bergamaschi et al. (2006). Henceforth, we refer to the data sets by the name of the first author (e.g., the Pollack data set). The Pollack and Bergamaschi data sets comprises of copy number and gene expression profiles, measured on cDNA microarray platforms, of 41 and 85 primary breast tumors (see Supplementary Material for pre-processing details). The gene expression of both data sets is used to investigate the link between the latent factors of a pathway's copy number data and its expression levels.
3.1. Characterization of the first two factors
First we investigate whether the interpretation of the two first EFA factors (the proportion of aberrations and the difference between the proportion of gains minus that of losses) as illustrated in Section 2.3 upholds more generally. Hereto we randomly selected 250 non-trivial (containing more than 20 gene and at most a 100 to exclude trivial and non-specific GO nodes, respectively) biological process category GO nodes. To the CNA data from each selected GO node, we fitted a two-factor model and estimated each sample's factor scores, its proportion of aberrations and its gain-vs-loss contrast. Spearman's rank correlations between the estimated factor scores and proportion and contrast for all GO nodes are calculated.
Figure 2 shows histograms of the absolute value of the Spearman's rank correlations. The upper panels show that the first EFA factor of a clear majority of GO nodes is highly correlated with the proportion of aberrations (Pollack: q0.25 = 0.88 and q0.50 = 0.93; Bergamaschi: q0.25 = 0.81 and q0.50 = 0.87). This observation does not come as a surprise. Bergamaschi and co-workers already noted that genome-wide the basal breast cancer subtype (as defined in Perou et al., 2000) is associated with a larger number of aberrations than other subtypes. Hence, as also the Pollack data set most likely (this information is not available) consists of all subtypes, one expects many GO nodes to reflect the genome-wide behavior: samples are distinguishable by their proportion of aberrations (the first EFA factor).
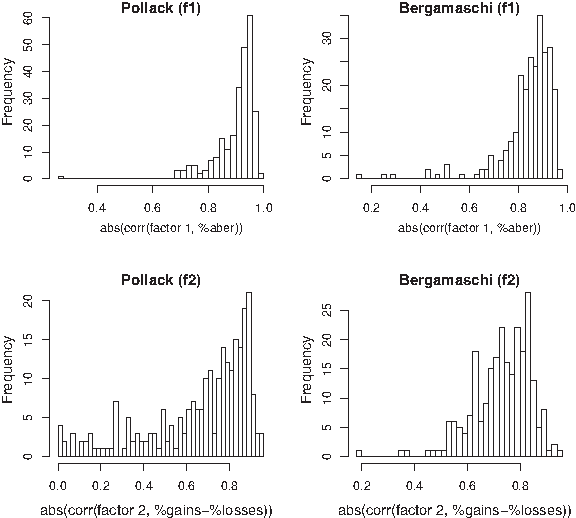
The lower panels of Figure 2 reveal that for a clear majority of the GO nodes the second EFA factor correlates with the contrast of gains and losses (Pollack: q0.25 = 0.61 and q0.50 = 0.78; Bergamaschi: q0.25 = 0.68 and q0.75 = 0.87). This contrast has not been noticed by Bergamaschi et al. (2006) as a characteristic separating the subtypes. In fact, Section 3.2 contains an example of a GO node where the latent factors reasonably separate the breast cancer subtypes basal from non-basal. Oddly enough, it is not the first factor, which correlates strongly with the proportion of aberrations, that is responsible for the separation, but the second factor, which correlates with the gain-vs-loss contrast. This is contrary to what was to be expected from the observation of Bergamaschi et al. (2006). EFA may thus give more subtle, specific information on a GO pathway's main characteristics than may be deduced from the genome-wide observation of Bergamaschi et al. (2006).
The above is of course no warrant that the two observed interpretations will always yield a good characterization of a pathway's copy number data variability. Associations may be of a weaker nature than observed here. In addition, the contrasts limited to a subset of the features in the pathway may yield a better association. This may also be the case for contrast between the percentages of non-gains and gains (or losses and non-losses), instead of between percentages of losses and gains. An example of a different interpretation than the above is given in Section 3.3, where the first factor is in fact correlated with the proportion of gains and the second with the proportion of losses.
3.2. Low-dimensional plotting
The latent variables may be used for low-dimensional plotting. In a plot, where each axis represents a latent variable, the estimated factor score vector of each sample is plotted. The samples are labeled in accordance with a clinical parameter. If the plot reveals that different values of the clinical parameter occupy different parts of the latent space, it seems possible to separate the samples with different clinical parameter values on the basis of the latent variables. An illustration is given in the left panel of Figure 3, where the latent factors reasonably separate the breast cancer subtypes (as defined in Perou et al., 2000) basal from non-basal. Another use of low dimensional plotting is to find corroboration of the sample subgroups found by (say) hierarchical clustering through an independent statistical technique, common practice in the analysis of gene expression data, for example, LaPointe et al. (2004). This is illustrated in the middle and right panel of Figure 3. The samples are clustered hierarchically using the average symmetric Kullback-Leibler divergence as a distance measure in combination with Ward's linkage (see Supplementary Material). The hard calls (instead of soft calls) are plotted in the heatmaps. The color bars above the heatmaps depict the two factor scores. Both factors are cut at their median and lower and upper 50% of the data are colored differently. This already indicates that the clustering may be explained by means of the latent factors, and is confirmed by the low dimensional plot in which the clusters separate reasonably well.
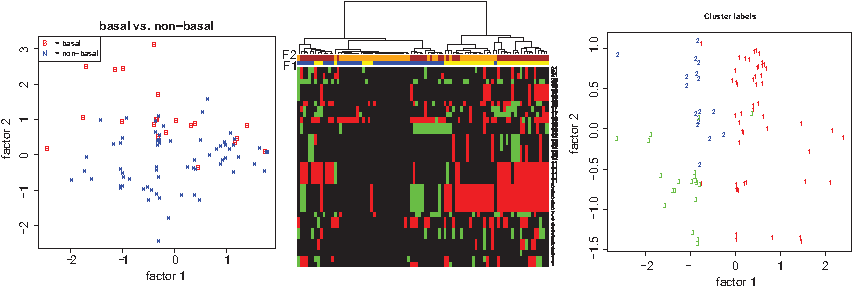
Heatmap and low-dimensional plots.
In principle the proposed method may be applied directly to copy number data from the whole genome (as opposed to that of a pathway), as is common in the analysis of gene expression data. We would then minimize the loss function over data from the whole genome. This practice may be acceptable as we are—in a strict sense—not working in a likelihood framework. It is also somewhat questionable as whole genome data are likely to violate the conditional independence assumption between features that is implicitly used in the motivation of the loss function. For there is a large redundancy in the data: many contiguous features have identical call probability signatures. Within these (possibly large) blocks of similar behaving features, the conditional independence assumption does not hold. An obvious way out would be to remove this redundancy by collapsing the data to the unique call probability signatures, for example, by a method described in Van de Wiel and Van Wieringen (2007). Application to in-house data sets shows promising results. Nonetheless care should be exercised when applying our method to the collapsed copy number data from the whole genome, as collapsing may not remove the spatial dependence fully.
3.3. Integration with gene expression
Pollack et al. (2002) claim a major direct role for CNAs in the transcriptional program. A re-analysis of their data set (Van Wieringen and Van de Wiel, 2009) confirmed that changes in expression levels of many genes are associated with gene dosage. We investigate whether this major direct effect observed in many genes individually upholds in pathways.
Hereto the factors of the 250 GO nodes are related to the gene expression data of the GO node. The association between the factors and gene expression is evaluated using GlobalANCOVA (Hummel et al., 2008). GlobalANCOVA is an ANOVA-based testing procedure that detects multivariate differential gene expression associated with a covariate of interest. Through GlobalANCOVA we thus investigate whether the EFA factors affect the multivariate gene expression levels in the pathway. Table 1 shows the results.
Number of significant GO nodes at various p-value cut-offs.
Table 1 indicates that both factors (in particular the first) are significantly associated with gene expression levels in many GO nodes. The fact that in the Pollack data set the number of GO nodes with a significant second factor falls behind that of the Bergamaschi data set may be merely a sample size effect (41 samples versus 85 samples). As with the common F-test that is able to detect smaller effect sizes with larger samples, the GlobalANCOVA F-test may benefit from more samples.
In order to assess whether this effect in GO nodes can be attributed to the “major direct effect” found in individual genes (Pollack et al., 2002), we calculated the residual gene expression, that is, gene expression corrected for copy number, from the mixture model relating copy number and gene expression proposed in Van Wieringen and Van de Wiel (2009) by means of a generalization of the method of moments approach used in Van Wieringen and Van de Wiel (2009) (see Supplementary Material). Using the residual gene expression data, we re-did the analysis above. Only a small decrease in the number of significant GO-nodes is observed. This suggests that, although the direct effect plays a major role, the factors have added value in many GO nodes. Put differently, the factors seem to capture a global (as opposed to direct) effect of copy number changes on gene expression.
We study two GO nodes in more detail to make the global effect of copy number of gene expression more intelligible. To the first, we fit a one-factor EFA model, and link its factor to the (original) gene expression data using GlobalANCOVA, which yields a p-value of 1.27 × 10−130. To explain the strong significance of the factor, the test statistic is decomposed into individual gene contributions. These contributions are directly related to the regression coefficients of the factor on the gene's expression levels. Closer inspection of the genes with a high contribution may help to understand the significance of the test. Although the factor is a representation of the percentage of aberrations aggregated over the genes in the GO node (ρ = − 0.95), it yields larger (absolute) correlations to the gene's expression levels than their individual percentages of aberrations (see Supplementary Material). The factor thus appears to contain more information on the expression of the genes in the GO node than their individual copy number signatures do. Finally, aggregation of the gene expression by averaging over genes in the GO node reveals a strong relation between the factor and the GO node's average gene expression (see Supplementary Material). A simple regression analysis reveals that the global copy number effect (exerted through the EFA factor) explains 53% (R2 = 0.53) of the average gene expression, where as the average R2 of the genes' individual regressions of gene expression on the proportion of aberrations equals R2 = 0.08 (minimum = 0.000, q0.25 = 0.012, q0.50 = 0.042, q0.75 = 0.094, maximum = 0.459). The adjusted p-values of these univariate regressions range from 2.7 · 10−10 to 1, of which 24 out of 91 are significant at an FDR cut-off of 0.05. To arrive at one p-value for the GO node the individual p-values may be combined into their geometric mean, as suggested by Fisher (1932), which would equal 0.034. Although significant at the α = 0.05 level, it does not come close to the GlobalANCOVA p-value for the EFA factor. This, together with a large difference in the R2, suggests that it is beneficial to aggregate a pathway's copy number data by means of EFA before testing the link with gene expression.
In another example, as shown in the upper panels of Figure 4, the first factor is associated with the percentage of gains whereas the second factor with the percentage of losses. Globally we expect both factors of this GO node to have an effect on the pathway's average gene expression, as is confirmed by GlobalANCOVA (p-value, Factor 1: 1.62 × 10−72; p-value, Factor 2: 5.27 × 10−12). In fact, both should have a positive effect as an increase of the number of gains as well as a decrease in the number of losses are expected to lead to a surge in transcription levels. The lower panels of Figure 4 confirm this for the first factor, but show only a weak positive correlation for the second. Indeed, a multivariate regression analysis returns positive coefficients for both factors, but only finds the first factor to have a significant association with the GO node's gene expression. A more in-depth univariate analysis of the GO node's 91 genes is needed to provide a better understanding of the significance of the second factor as found by GlobalANCOVA.
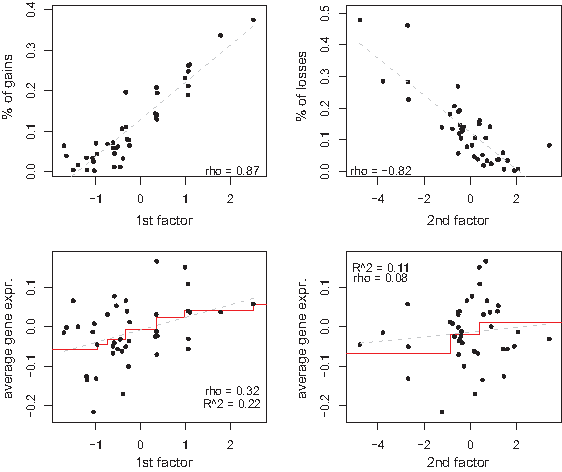
Others (Ferreira et al., 2008; Järvinen et al., 2008; Lee et al., 2008; Van Wieringen and Van de Wiel, 2009) have also studied copy number and gene expression data jointly within pathways. The analyses in these articles all boil down to a univariate analysis (which genes show an association between copy number and gene expression levels), followed by a gene set enrichment procedure (Subramanian et al., 2005) revealing pathways with an excess of genes with an association between the molecular levels. Although an obvious approach, it ignores much of the complexity of the data. We do not claim to have resolved this complexity with the analysis presented above. But as EFA captures the key characteristics of the pathway copy number data through a multivariate analysis, the key characteristics may be more helpful to explore the complexity of data from the two molecular levels within pathways.
3.4. Comparison
The exploratory factor analysis results of a GO node's call probability data are compared with principal component analysis of the normalized and the segmented array CGH data, as is done by, for example, Somiari et al. (2004) and Unger et al. (2008). To this end, we take the set of 250 GO nodes used previously. For each GO node, we calculate the first principal component (corresponding to the largest eigenvalue) of both the normalized and segmented data. These are used to calculate the Spearman rank correlation coefficient between the principal components and sum of the absolute log2 ratios of the genes in the GO node. The latter is a proxy for the proportion of aberrations. We also test the association between the components and the GO node's multivariate expression using GlobalANCOVA. These results are compared to the results of the analyses in Sections 3.1 and 3.3. Figure S5 in the Supplementary Material displays the comparison between the results of EFA and PCA on the segmented data.
The figure shows that the factors constructed with the EFA tend to be higher correlated with the proportion of aberrations than the principal components of the segmented data. The figure also reveals that the factors are more significantly associated with the gene expression than these principal components. PCA of normalized array CGH performed worse than both EFA and PCA with segmented data (results not shown). Overall we conclude that EFA is to be preferred over the PCA of either normalized or segmented data.
Besides these quantitative arguments, there is also an important qualitative argument in favor of EFA. Exploratory statistical techniques like EFA and PCA aim to identify salient features of the data. Interpretation of the identified salient features may lead to the generation of new hypotheses regarding the phenomenon under study. It is this interpretation that EFA factors naturally inherit from the call probabilities, and which facilitates understanding and hypothesis generation. The interpretation of the PCA principal components is much harder, see for instance our use of a proxy for the proportion of aberrations in the quantitative comparison above.
4. Conclusion
We presented EFA to characterize the variability in a pathway's copy number data. The proposed method introduces latent variables to model this variability and uses an EM algorithm to fit the model. Practical guidelines as how to assign meaning to the latent variables are given. The potential of our exploratory factor analysis was illustrated in two breast cancer data sets, where it identified several salient features of the studied GO nodes' copy number data. Among others the method suggested that the proportion of aberrations is a key characteristic of a pathway's CNA pattern. It also suggested that another key characteristic may be found in gain-vs-loss contrasts, which should possibly be limited to a subset of genes in the pathway. These clear interpretations stem from the fact that EFA analyzes copy number data represented as call probabilities, and are not possible with convential PCA. In the GO nodes studied, the EFA characteristics were often significantly associated with the node's gene expression data, and may be considered to capture the pathway's global effect of copy number changes on transcription levels. In particular, it yields a better association with gene expression (in terms of R2 and p-value) than univariately based approaches that have employed to analyze data from the two platforms. Finally, the results of EFA may support results from hierarchical clustering and be useful for low dimensional plotting. In all, we believe that EFA will prove to be a fruitful technique for exploratory analysis of pathway copy number data.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.