
Abstract
Abstract
The problem of inferring phylogenies (phylogenetic trees) is one of the main problems in computational biology. There are three main methods for inferring phylogenies—Maximum Parsimony (MP), Distance Matrix (DM) and Maximum Likelihood (ML), of which the MP method is the most well-studied and popular method. In the MP method the optimization criterion is the number of substitutions of the nucleotides computed by the differences in the investigated nucleotide sequences. However, the MP method is often criticized as it only counts the substitutions observable at the current time and all the unobservable substitutions that really occur in the evolutionary history are omitted. In order to take into account the unobservable substitutions, some substitution models have been established and they are now widely used in the DM and ML methods but these substitution models cannot be used within the classical MP method. Recently the authors proposed a probability representation model for phylogenetic trees and the reconstructed trees in this model are called probability phylogenetic trees. One of the advantages of the probability representation model is that it can include a substitution model to infer phylogenetic trees based on the MP principle. In this paper we explain how to use a substitution model in the reconstruction of probability phylogenetic trees and show the advantage of this approach with examples.
1. Introduction
The reconstruction of phylogenetic trees is based on nucleotide or protein sequences (bio-sequences). In this article, the sequences we study are uncoded nucleotide sequences that consist of characters from the alphabet {A,C,G,T} but all the arguments can be applied to proteins or other kind of sequences. Of all the methods developed for phylogenies—such as Maximum Parsimony (MP), Distance Matrix (DM), and Maximum Likelihood (ML)—the MP method is the most well- studied and popular method because of its simplicity. The principle of Maximum Parsimony is like Ockham's razor: in the absence of contrary information, the simplest is the best, i.e., the simplest manipulations of the observed data are the best explanation for the data. In the case of inferring phylogenies, this principle can be described as “evolution is parsimonious.” Specifically, the amount of evolutionary change (the total number of substitutions of nucleotides) is minimized. The MP method relies on directly observable changes in the input bio-sequences. Although the MP method continues to be widely used, it is often criticized as not being statistically sound, i.e., there is a lack of assumption involving an underlying substitution model (Steel, 2000). The reason for this is that the actual substitution patterns are very complicated: In the evolutionary process there exist multi-, parallel-, convergent-, and back-substitutions (Xia, 2006). Multi-substitutions result in the number of observable substitutions being less than the actual occurring multi-substitutions, while the last three kinds of substitutions mentioned above cannot be observed at all in the comparison of sequences. The cost of the simplicity of the MP method is that these unobserved substitutions are ignored and the number of observable substitutions typically greatly underestimates the actual number of substitutions in the evolutionary history. In order to reflect these invisible substitutions, a number of substitution models such as the Jukes-Cantor model (Jukes et al., 1969) have been developed (Ewens et al., 2005). These substitution models are now widely used in the DM and ML methods. However, because the MP method is character-based and because it cannot take into account different possible substitutions on an edge simultaneously, the substitution models cannot be applied in the classical MP method.
The existing MP method is deterministic in the sense that any internal node at a site is determined as an “either this or that” nucleotide. Because an evolutionary history cannot be repeated, we are unable to know exactly the ancestors of a given set of organisms. Hence, a more logical approach is to assume that each site of an internal node is a probability distribution of the four types of nucleotide. For instance, if the distribution on a site of a node is [A,C,G,T] = [0.80, 0.18, 0.01, 0.01], then we can say that during the evolutionary history the nucleotide on this site was most likely to be A, with a small probability it was C, but it was most unlikely to be G or T. Based on this argument the authors have recently proposed a probability presentation model of phylogenetic trees and the trees constructed in this model are called probability phylogenetic trees (Weng et al., 2008). The probability representation model makes it possible to use a substitution model in reconstructing probability phylogenetic trees by the MP principle. In this article, we explain this new approach and, using examples, show the advantage of the proposed approach over the classical MP method.
2. Probability Phylogenetic Model and Statistical Distance
The problem of reconstructing phylogenies is closely related to the Steiner tree problem, a well-studied problem in combinatorial optimization (Foulds et al., 1982; Brazil et al., 2009). The classical Steiner tree problem has two versions: in graphs or in metric spaces. The probability representation model is related to the real d-dimensional space version whose mathematical formulation is as follows:
is minimized, where E(T) is the edge set of T, w
Due to minimality, the optimal network T does not contain cycles. That is, T has a tree topology, and hence, T is called a Steiner minimal tree. There are two “levels” of optimisation of T in the Steiner tree problem. T is locally optimal if it is optimal with respect to a fixed topology, i.e., optimal over all networks with the same topology, while T is globally optimal if it is optimal over all feasible topologies. In this article, our focus is mainly on local optimisation although the comparison of topologies on the global optimisation level is also discussed in Example 3 in Section 4.
A vector
Note that when a sequence is produced in a wet laboratory, it may contain uncertain characters (ambiguous characters). Besides, gaps may occur due to insertion/deletion of characters in alignment. Moreover, in the reconstruction of phylogenetic trees using the MP method, an optimal assignment of internal nodes on each site, called an evolutionary pathway on the site, is often not unique. Due to this non-uniqueness, for a fixed site the length of an edge is defined to be the average of the numbers of substitutions over all evolutionary pathways. The ambiguity in the input sequences and the non-uniqueness of the output have lead us to consider the probability presentation model. More importantly, as we have stated in the first section, the evolutionary history of a given set of species is not repeatable and we cannot precisely infer the ancestors of the species. Hence, instead of a determinate assignment of nucleotides to an internal node in pathways, in the probability representation model of phylogenetic trees a distribution of the nucleotide states will be inferred for each site of each internal node. The probability representation model consists of two steps:
converting all nucleotide sequences in the input set N into probability vector sequences, consequently the input becomes a set N of probability matrices, and constructing a probability phylogenetic tree T that is actually a probability Steiner minimal tree spanning the input set N of probability matrices.
Let T be the phylogenetic tree on a set N of n nucleotide sequences of equal length m, and T
k
be the tree constrained to site k. First we map all characters (nucleotides or non-nucleotide characters) in the input sequences to points in a 4-dimensional probability vector space
All other non-nucleotide characters (uncertain characters and gaps) need to be determined using particular methods. For example, in Example 2 (see Section 4 below) the character on site 6 in the sequence of Turkey is “N,” a character in the table of the IUPAC-IUB Commission on Biochemical Nomenclature (Liébecq, 1992), meaning an A, or C, or G, or T, i.e. any nucleotide is possible. Therefore, we will assume the probability distribution is [0.25,0.25,0.25,0.25] if no bias is given beforehand. Once the distributions of all non-nucleotide characters are determined, then a nucleotide sequence
such that each column
Remark 2.1.
(1) In the notation (2) We use (3) For n input sequences each site can be treated as an n × 4 probability matrix in which each row is a probability vector. An example is given in Section 4.
After the input set N of nucleotide sequences is translated into a set N of 4 × m probability matrices, in the second step of the probability representation model, a proper length measure of edges, i.e., a distance measure between the endpoints of edges, needs to be defined. That is, the measure of an edge
Remark 2.2.
Here ‘length’ or ‘distance’ is not a geometric term but used as a general term measuring the difference between sequences or probability distributions. Therefore, the triangle inequality does not necessarily hold.
Once the function fℓ is defined, the original phylogenetic tree problem is then transformed into a probability-constrained Steiner tree problem on N and we can use existing optimisation programs to solve it (Liberti et al., 2006).
Now we elaborate on the MP method using the probability representation model. The optimisation criterion in MP is the total number of differences between the corresponding characters in the input sequences. For two character sequences P and Q the number of differences between P and Q is known as the Hamming distance in the literature (Althaus et al., 2006) but referred to as seq-difference and denoted by ndiff(P,Q) in this article:
The continuous generalization of the Hamming distance in our model is statistical distance (i.e., total variation distance. For two probability distributions
For example, the difference between two different nucleotides, say A and C, is one, which is just the statistical distance between their mappings, i.e. two probability distributions [1,0,0,0] and [0,1,0,0] is given by:
Hence, from the correspondence p
i
⇔
The following theorems describe two basic properties of probability vectors and the statistical distance in general d-dimensional vector space.
Theorem 2.1.
For any two probability vectors
Suppose the statement holds for n ≥ 1 and suppose now d = n + 1. Note that there are at least two components, say the first two components, satisfying p1 ≥ q1, p2 ≥ q2, or vice versa. Assume the former case. Let
Theorem 2.2.
Suppose
3. A Substitution Model for Inferring Probability Phylogenetic Trees
The number of substitutions between two sequences per site is known as a genetic distance. The simplest genetic distance, known as the p-distance dp, between two nucleotide sequences P, Q, is the ratio of the total number of differences ndiff(P,Q) divided by the sequence length m:
Because MP is site based, we can omit the factor 1/m and simply let m = 1, then
As we stated in the first section, the p-distance typically much underestimates the actual number of substitutions in the evolutionary history, and hence a substitution model is needed to correct. All the existing substitution models are based on a continuous Markov process that describes how a probability distribution
In phylogeny, Q is the matrix whose entries are the instantaneous change rates of the nucleotides. In inferring phylogenies because we have four types of nucleotides (or in other words, four states of nucleotides) Q is a 4 × 4 matrix whose entry q
i,j
is the instantaneous rate of changes from state i to state j. In the simplest model, called the Jukes-Cantor model (JC69-model) (Jukes et al., 1996), all four types of nucleotide have the same instantaneous rate α of change, i.e.,
The solution of Equation (6) is
The matrix
Let μ be the rate of substitution on an edge
Note that in this equation dJC is a function of the original statistical distance dstat, and the domain of the variable is 0 ≤ dstat ≤ 3/4. This is guaranteed by Theorem 2.2. The curve of the function dJC, as a function of dstat, is depicted in Figure 1.
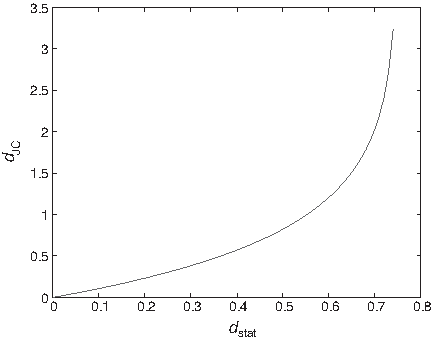
dJC, as a function of dstat.
A statistical model is currently applied to sequences in the Distance Matrix method (DM) or to characters as in the Maximum Likelihood method (ML). Now we apply the JC69-model to the probability representation model so that for two probability vectors
Using this corrected statistical distance dJC the MP approach for inferring probability phylogenetic trees becomes a problem asking for a probability Steiner tree T spanning a given set of probability matrices such that the total tree length
is minimized.
Theorem 3.1.
dJC is convex.
This is equivalent to prove
or
Because ℓ1 is convex,
Hence the left side of Equation (12) is
The theorem is proved.
By this theorem L(T) in Equation (11) is convex and the phylogenetic tree problem for a given topology becomes a convex optimisation problem. Therefore we can use a convex optimisation program, say CVX (Grant et al., 2009), to solve it. Here we must point out that dJC is not a real metric in a mathematical sense because it does not satisfy the Triangle Inequality. But the above formulation of the phylogenetic tree problem does make sense because the following theorem holds.
Theorem 3.2.
The set of feasible solutions of the Steiner points in T under the JC69-distance is not empty.
Let
Let
Let
be three randomly generated probability vectors. Then the tree length L(T) with respect to the Steiner points in Δ

L(T) with respect to the Steiner points p + x(q − p) + y(r − p) in Δpqr.
4. Two Examples
Example 1.
Below is a simple example that consists of 5 sequences of 42 nucleotides (Felsenstein, 2009).
Turkey AAGCTNGGGC ATTTCAGGGT GAGCCCGGGC AATACAGGGT AT Salmo gair AAGCCTTGGC AGTGCAGGGT GAGCCGTGGC CGGGCACGGT AT H. Sapiens ACCGGTTGGC CGTTCAGGGT ACAGGTTGGC CGTTCAGGGT AA Chimp AAACCCTTGC CGTTACGCTT AAACCGAGGC CGGGACACTC AT Gorilla AAACCCTTGC CGGTACGCTT AAACCATTGC CGGTACGCTT AA
Note that there is an uncertain character “N” on site 6 in the sequence of Turkey, therefore in the probability representation model the input sequences on site six, [NTTCC]
T
, will be represented by a 5 × 4 probability matrix
Let TMP be the classical phylogenetic tree inferred by the classical MP method (e.g., Fitch's algorithm) (Fitch, 1971; Swofford et al., 1996), and Tp and TJC be the optimal probability phylogenetic trees inferred using the standard statistical distance and JC69-distance respectively. First we should explain the difference caused by the different treatments of the character “N” in the classical MP method and in the probability representation model. In inferring TMP using the classical MP method the character “N” is treated as a “wild card,” therefore, no substitution is incurred when “N” is compared with any other nucleotide character. However, as stated above, in the probability representation model ‘N’ is a probability vector with equal distributions at each component. As a result, L(Tp) = 44.7497 is larger than L(TMP) = 44.
Remark 4.1.
As a comparison, assume that the character on site 6 in Turkey is not “N” but a gap “-.” Because in the classical MP method a gap is regarded as a 5th character different from A, C, G, T, and treated as neither A, nor C, nor G, nor T, one substitution is definitely incurred and the tree length TMP would become 45.
The two probability phylogenetic trees Tp and TJC are shown in Figure 3. From the picture it is clearly seen that L(TJC) is much larger than L(Tp) (is about the double of L(Tp)). This is because the unobservable changes of nucleotides are taken into account in TJC but are not in Tp.

Two probability phylogenetic trees of Example 1.
Example 2.
This is a real example: The given set N containing 14 species is a part of a group of 25 species, originally studied in (Stanhope et al., 1996). The 14 sequences in N, listed in the Appendix, are 532 nucleotides long. Four different probability phylogenetic trees on N are shown in Figure 4 and the differences between the four probability phylogenetic trees are summarized in Table 2.
T1: The tree is inferred by the MP method using the statistical distance dstat. Because there are no ambiguous characters in the input sequences its topology topoMP and the tree length L(T1) = 731 is the same as the optimal phylogenetic tree reconstructed by a classical MP method (Weng et al., 2008). T2: The tree is inferred with the same topology as T1 but the length function is the JC69-distance dJC. Note that, as in Example 1, we can see again that L(T2) = 1428.66, is much larger than L(T1) = 731 and L(T2)/L(T1) ≈ 2. T3: As mentioned above the Distance Matrix methods such as Neighbor Joining (NJ) can use substitution models to estimate unobservable substitutions. Let topoNJ+JC denote the topology of the classical phylogenetic tree that is inferred by NJ using the JC69-model. Then T3 is the probability phylogenetic tree that is inferred using JC69-distance with the topology topoNJ+JC. L(T3) = 1424.66, smaller than L(T2). T4: Whether a tree is globally optimal or not depends on its topology. For this reason, T3 is not the optimal probability phylogenetic tree because T4 has a better topology and L(T4) = 1424.55, a little smaller than L(T3). In this example we didn't search the whole topology space of probability phylogenetic trees, hence, the topology of T4 is possibly only sub-optimal, and denoted by toposubopt.

Four probability phylogenetic trees of Example 2.
5. Conclusion
We have laid out a new approach to the reconstruction of phylogenetic trees that combines the probability representation model with a substitution model. From the demonstrated examples, we can see that the probability representation model gives a solution to the puzzle of ambiguity and non-uniqueness in the reconstruction of phylogenetic trees. This probability representation model, which includes a substitution model, yields topologies, edge lengths, and ancestor states that are more likely to represent real biological evolution than that where no substitution model is used.
6. Appendix
Fourteen species of mammals
1. Marsupial Mole
GCTCCAGCAAATGATCAAGTACCAGGTATTGGAGGGCAATGTGGGTTACCTAAGAGTGGACTACATCCCTGGCCAG GAGGTAGTAGAAAAAGTCGGGGAGTTCCTGGTGAATGACATCTGGAAGAAGCTCATGGGGACATCCTCTCTAGTGC TAGATCTCCAGCACAGCACAGGGGGTGAAGTTTCGGGAATCCCCTTTGTCATTTCCTATCTACATCAGGGGGATAT CCTGCTCCATGTAGACACAGTTTATGACCGGCCATCAAACACTACCACAGAGATCTGGACCCAGCCTCAGGTGCTG GGTGAGAGGTATGGAGGGGAGAAGGACATGGTGGTTCTCACCAGCCATCATACTGTAGGGGTAGCTGAGGATATCG CCTATATTCTCAAGAAGATGCGCCGGGCCATTGTGGTGGGAGAGCAGACTCTGGGAGGGGCCCTAGATCTCCGGAA GCTGCGCATCGGTCAGTCAGACTTTTTCATCACTGTGCCCGTGTCACGCTCCCTGAGCCCCCTTGGTGGGGGGAGT
2. Wombat
GCTCCAGCAAATGATCAAGTACCAGGTACTGGAGGGTAATGTGGGTTACCTGAGAGTGGACTACATCCCTGGCCAG GAGGTGGTAGAGAAAGTCGGGGAGTTCCTGGTGAATGATGTCTGGAAGAAGCTCATGGGGACCTCTTCTCTGGTGT TGGATCTCCAGCACAGCACGGGAGGCGAAGTTTCAGGAATCCCGTTTGTCATTTCCTACCTACACCAGGGGGATAA TCTGCTGCATGTAGACACAGTTTATGACCGGCCATCAAACACCACCACAGAGATCTGGACCCTGCCCCAGGTGTTG GGTGAGAGGTACGGTGGGGAGAAGGACGTGGTGGTCCTCACCAGCCATCACACGGTCGGGGTAGCAGAGGATATTG CCTACATCCTCAAGAAGATGCGCCGGGCCATTGTGGTGGGAGAGCAGACTCTGGGAGGGGCCCTAGATCTCCGGAA GCTTCGTATTGGTCAGTCAGACTTTTTCATCACTGTGCCCGTGTCCCGTTCTCTGAGCCCCCTCAGTGGGGGGAGC
3. Rodent
GCTACAGAGGAATATTCACCATGAGGTTCTGGAGGGCAACTTGGGTTACCTATGGGTGGACGATCTCTTGGGCCAG GAGGTACTGAGTAAGCTCGGGGGATTCCTGGTGGCCCACATGTGGGGGCAGCTCATGAATACCTCTGGCTTGGTGC TAGATCTCCGGCACTGTACTGGGGGGCATGTTTCTGGTATTCCCTATGTCATCTCCTACTTGCACCCCGGGAACAC AATCATGCATGTGAACACCATCTATGATCGGCCCTCTAATACCACCACAGAGATCTGGACCTTGGCCAAGGTCCTG GGGGAGAGGTACAGTGCTGACAAGGATGTGGTGGTCCTCACCAGTGGCCACACTGGAGGAGTGGGTGAGGACATTG CCTATATCCTCAAACAGATGCGCAGGGCCATCATGGTGGGTGAGCAGACTGAAGGTGGTGCCCTGGACCTCCAGAA ACTGAGGATAGGCCAGTCCAACTTCTTCCTCACAGTGCCTCTGGCGATGTCTCTGGGGCCGATGGGTGGAGGTGGC
4. Elephant Shrew
GCTGGAGAGAAGCATGAGCTACAGGATTCTGGATGGTAATGTGGGCTACTTGCAGATAGACAACATCCCAGGCCAG GAGGTACTGAGCCGACTAGGGGCCTTCCTGGTGGCCCATGTCTGGAGACAGCTCATGGGCACCTCTGCTTTGGTGT TGGACCTGCGGCAGTGCACAGGAGGCCATGTTTCCAGCATCCCTTACCTTATTTCCTACCTGCACCCAGCGGGCAC GGTCCTGCACGTTGACACCATTTACAACCGTCCCTCTAACACAACCACTGAGCTCTGGACTTTGCCTCAGGTGCTT GGGGAGAGATACAGTGCTGAGAAGGATGTGGTGGTCCTCACCAGTGGTCAAACCCGGGGTGTGGCTGAGGACATTG TCTACATCCTCAAGCAGATGGGCAGGGCCATAGTGGTGGGTGAACGTACTGGGGGGGTCTCCCTGGACCTCCAGAA GCTAAGGATAGCCAACTCTGACTTCTTCCTCACTCTACCTGTGTCCAGGTCCTTGGGGCCTCTGGGTGGAGGCACC
5. Elephant
GCTGCAGACAAGCATGAGCTACAAGGTTCTGGAGGGCAACGTGGGCTACCTGCGGGTAGACAACATCCCAGGCCAG GAGGTGCTGAACCAGCTGGGGGCCTTCCTGGTGACTCACGTCTGGAAGCAGCTTATGGGCTCCTCTGCCTTAGTGC TGGACCTGCGACACTGCACAGGGGGCCATGTCTCCAGCATCCCTTACCTCATTTCCTACCTGCACCCGGGCGGCAC CGTGCTGCACGTGGACACCATTTACAACCGCCCCTCCAATACGACTACGGAGCTCTGGACCTTGCCCCAGGTGCTG GGGGAGAGGTATAGCGCCGACAAGGATGTGGTGGTCCTCACCAGTGGCCACACCAGGGGCGTGGCCGAGGACATCG TCTACATCCTCAAGCAGATGGGCAGGGCCATCGTGGTGGGCGAGCGGACTGAGGGTGGTGCCCTGGACCTCCAGAA GATAGGCCACTCTGACTTCTTCCTCACTCTGCCTGTGTCTAGGTCCTTAGGCCCCCTGGGCGGGGGAAGCCAGACA
6. Whale
GCTGCAGAACGGCCTCCGCCATGAGGTTCTGGAAGGCAATGTGGGCTACCTGCGGGTGGACGACATCCCAGGCCAG GAGGTGATGAGCAAGCTGAGGAGCTTCCTGGTGGCCAACGTCTGGAGGAAGCTCATGGGCACCTCTGCCTTGGTGC TGGACCTCCGCCATTGCACTGGGGGCCACATTTCTGGCATCCCCTATGTCATCTCCTACCTGCACCCGGGGAACAC AGTCCTGCACGTGGATACCATCTATGATCGCCCCTCTAATACGACCACTGAGATCTGGACCCTGCCCGAAGTCCTA GGAGAGAACTACGGTGCCGATAAGGATGTGGTGGTCCTCACCAGTGGTCGCACCGGGGGTGTGGCTGAGGACATCG CTTATATCCTCAAACAAATGCGCAGGGCCATTGTGGTGGGCGAGCGGACTGTGGGGGGGGCCTTGGACCTCCAGAA GATAGGCCAGTCTGACTTCTTTCTCACCGTGCCCGTGTCCAGGTCCCTGGGGCCCCTGGGCAAGGGCAGTCAGACT
7. Dolphin
GCTGCAGAACGGCTTCCGCCATGAGGTTCTGGAAGGCAATGTGGGCTACCTGCGGGTGGACGACATCCCGGGCCAG GAGGTGATGAGCAAGCTGAGGAGCTTCCTGGCGGCCAACGTCTGGAGGAAGCTCATGGGCACCTCTGCCTTGGTGC TGGACCTCCGCCACTGCACTGGCGGCCACATTTCCGGCATCCCCTATGTCATCTCCTACCTGCACCCAGGGAACAC AGTCCTGCATGTGGATACCATCTACGATCGCCCCTCTAATACGACCACTGAGATCTGGACCCTCCCCGAAGTCCTA GGAGACAACTACGGTGCCGATAAGGATGTGGTGGTCCTCACCAGTGGTCGCACGGGGGGTGTGGCTGAGGACATCT CTTATATCCTCAAACAGATGGACAGGGCCATCGTGGTGGACGAACGGACTGTGGGGGGGGCCTTGGACCTCCAGAA GATAGGCCAGTCTGAGTTCTTTCTCACAGTGCCCGTGTCCAGGTCCCTGGGGCCCCTGGGCAAGGGCAGCCAGACT
8. Pig
GCTGCACAATAGTCTCCGCCATGAGGTTCTGGAAGGCAATGTGGGCTACCTGCGGGTGGACGACATCCCAGGCCAG GAGGTGATGAACAAGCTGGGGAGCTTCCTGGTAGTCAACGTCTGGGAAAAGCTAATGGGCACCTCTGCCTTGGTGC TAGACCTCCGGCACTGCACCAGGGGCCACGTTTCTGGCATCCCCTATGTCATCTCCTACCTGCACCCAGGGAACAC GGTCCTGCACGTGGACACCATCTATGACCGTCCCTCCAATACGACCACTGAGATCTGGACCCTGCCCGAAGTCCTG GGAGACAGGTACAGTGCGGATAAGGACGTGGTGGTCCTCACCAGCAGCCACACAGGGGGCGTGGCTGAGGACATCG CCTACATCCTCAAACAGATGCGCAGGGCCATTGTGGTCGGCGAGCGAACTGTGGGGGGTGCCCTGGACCTCCAGAA GATAGGCCAGTCCGACTTCTTTCTCACCGTGCCTGTGTCCAGGTCCCTGGGGCCCCTGGGTGAGGGCAGCCAGACA
9. Horse
GCTGCAGGAGGGCATCCGCTATGACATTCTGGAGGGCGACGTGGGCTACTTGCGAGTGGACAACATCCCGGGCCAG GAGGTGGTGAGCAAGCTGGGGGGCTTCCTGGTGGACAATGTCTGGAGGAAGCTCATGGGCACCTCTGCCTTGGTGC TGGACCTCCGGCACTGCACTGGGGGCCACGTTTCCGGCATCCCCTATATCATCTCCTACCTGCACCCAGGAAACAC GGTCCTGCACGTGGACACCATCTACGACCGCCCCTCCAATACGACCACTGAGATCTGGACCCTGCCCGAGGTCCTG GGAGAGAGGTACAGTGCCGACAGGGATGTGGTGGTCCTCACCAGTGGCCACACCGGGGGCGTGGCCGAGGACATTG CTTACATCCTCAAACAGATGCGCAGGACCATCGTGGTGGGTGAGCGGACCGTGGGAGGTGCCCTGGACCTCCAGAA GATAGGCCAGTCCGACTTCTTCCTCACCGTGCCCGTGTCCAGGTCCCTGGGTCTGCGCGAGGTCCTCATGCATAAC
10. Bat
GCTGCAAAAGGCCATCCACTACAATGTTCTGGAGGGCAACGTGGGCTACTTTCGGGTGGACGACATCCCGAGCCAG GAGGTGGTGAGCAATCTTGGGGGCTTCCTCGTGGACAATTTCTGGAGGAAGCTCCTGGGCACCTCTGCCTTGGTGC TAGACCTCCCACACTGCACTGGGGGGCACGTTTCTGGGATCTCCTATGTCATCTCCTACTTGCACCGAGGGAACAC CGTCCTGAATGTGGACCCACTCTATGACCCCCCCTCCAACACGACCACAGAGATCTGGACCCTGCCCCAGGTCCTG GGAGAGAGGTACAGTGCTGACAAGGATGTTGTGGTCCTCACCAGTGGCCACACTGGAGGAGTGGCTGAGGACATTG CTTACATCCTCAAACAGATGCGCAGGGCCATTGTGGTGGGTGAGCAGACTGTGGGGGGTGCCCTGGACCTCCAGAA GATAGGCCAGTCTGACTTCTTCCTCACTGTGCCTGTGTC TAGGTCCCTGGGGGCTCTGGGTGGGGGCAGGCAGACA
11. Insectivore
GCTGCAGAGGGCCATCCGCTACCAGGTTCTGGCGGCCAATGTGGGCTACCTGGGGAGGGATAACCTCCCCGGTCAG GAGGTGGTGACCATACTGGGGGCTCTCCTGGTGGCCAATGTCTGGGGGAAGCTCATAGCCACCTCTCCCTTGGTGC TGGACCTCCGACACTGCACTGGGGGCCATGTCTCTGGGATCCCCTACGTCATCTCCTACCTGTACCCAGGAAACAC GGTCCTGCATATGGACACCATCTATGACCGCCCCTCCAATATCACCACTGAGCTCTGGACCCTGCCCCAGCTCCAG GGAGAGCGGTACGGTGCAGACAAGGATGTGGTGGTCCTCATCAGCGACCACACTGGGGGTGTGGCTGAGGACATTA CTTACATCCTCAAACAGATGCGCCGGGCTATTGTGGTGGGCGAGCAGACTGTGGGGGCTGCTCTGGACCTCCAGAA GATAGGCCAGTCTGACTTCTTCATCACTCTGCCTGTCTCCAGGTCTCTGGGGACTCTGGGCGGGGGCAGCCAGACA
12. Human
GCTGCAAAGGGGCCTCCGCCATGAGGTTCTGGAGGGTAATGTGGGCTACCTGCGGGTGGACAGCGTCCCGGGCCAG GAGGTGCTGAGCATGATGGGGGAGTTCCTGGTGGCCCACGTGTGGGGGAATCTCATGGGCACCTCCGCCTTAGTGC TGGATCTCCGGCACTGCACAGGAGGCCAGGTCTCTGGCATTCCCTACATCATCTCCTACCTGCACCCAGGGAACAC CATCCTGCACGTGGACACTATCTACAACCGCCCCTCCAACACCACCACGGAGATCTGGACCTTGCCCCAGGTCCTG GGAGAAAGGTACGGTGCCGACAAGGATGTGGTGGTCCTCACCAGCAGCCAGACCAGGGGCGTGGCCGAGGACATCG CGCACATCCTTAAGCAGATGCGCAGGGCCATCGTGGTGGGCGAGCGGACTGGGGGAGGGGCCCTGGACCTCCGGAA GATAGGCGAGTCTGACTTCTTCTTCACGGTGCCCGTGTCCAGGTCCCTGGGGCCCCTTGGTGGAGGCAGCCAGACG
13. Sea Cow
GCTGCAGACCAGCATGAGCTACAAGGTTCTGGATGGCAATGTGGGCTACCTGCGGGTAGACAACATCCCTGGCCAG GAGGTGCTGAGCCGTCTGGGGGGCTTCCTGGTGACTCACATCTGGAAGCAGCTCATGGGCTCCTCTGCCTTAGTCC TGGACCTGCGGCACTGTATGGGTGGCCATGTCTCCAGCATCCCTTACATCATCTCCTACCTACACCCCGGAGGAGC AGTGCTGCATGTGGACACCATTTACAACCGCCCCTCCAATACGACTACTGGGGTCTGGACCTTGCCCCAAGTGCTG GGAGAAAGGTACAGTCCCAACAAGGATGTGGTGGTCCTCACCAGTGGCCACACCAGGGGCGTGGCCGAAGACATCG TTCACATCCTTAAGCAGATGGGCAGGGCCATAGTGGTGGGCGAGAAGACGGAGGCAGGTGCCCTGCACCTCCAGAA GATAGGTCACTCTGATTTCTTTCTCACTCTGCCTGTGTCCAGGTCCTTGGGGCCTTTGGGCAGGGGAAGCCAGACA
14. Hyrax
ACTGCAGACAAGCATGAGCTACAAGGTTCTGGAGGGCAACGTGGGTTACCTGCGGGTAGACAACATTCCGGGTCAA GATGTGCTGAACCAGCTGGGGGGCTTCCTGGTGACTCATGTGTGGAAGCAGCTCATGGGCTCCTCTGCCTTAGTGC TGGACCTAAGGCACTGCACGGGGGGCCATGTCTCCAGTATCCCTTACCTCATCTCCTACCTGCATCCAGGGAGCAC TGTGCTGCACGTGGACACCATTTACAACCGCCCCTCCAATACAACTACTGAGCTCTGGACCTTGCCCCAGGTGCTG GGGGAGAGATACAGTGCTGACAAGGATGTGGTGGTCCTCACCATGGGCCACACCAGGGGTGTGGCCGAGGACATCG TCTACATCCTCAAGCAGATGGGCAGGGCCATTGTGGTAGGCGAGCGGACCGAGGGTGGTGCCCTGGACCTCCAGAA AATAGGTCACTCAGACTTCTTTTTCACTCTGCCTGTGTCCAGGTCACTGGGCCCCTTAGGCAGGGGAAGCCAGACA
Footnotes
Disclosure Statement
No competing financial interests exist.