
Abstract
Abstract
1. Introduction
Proteins, for example, are optimized by selection to assume specific chemical properties and spatial conformations: this streamlining tends to remove redundancies (Anfinsen, 1972), yielding strings of amino acids in which every symbol carries information; such “slightly edited random strings” (Weiss et al., 2000) are therefore hardly distinguishable from their random permutations when measured with both statistical and algorithmic definitions of information. Not even translating amino acids with scales that capture relevant physico-chemical properties provides significantly more insight: for example, the distribution of hydrophobicity—a key property influencing folding and spatial stability—along the sequence of most proteins is well known to be indistinguishable from random (White and Jacobs, 1990; Schwartz and King, 2006). The very presence of repetitions and redundancies has been implicated in human diseases at the DNA level (Benson and Waterman, 1994) and in the formation of toxic fibrillar structures at the protein level (Broome and Hecht, 2000). Repetitions in polypeptides have also been conjectured to multiply the folding possibilities by introducing many interactions with similar energy (Wan and Wootton, 2000): these alternatives would prevent the convergence of the folding process into a global minimum. Wet-lab experiments with random polypeptides (Ptitsyn and Volkenstein, 1986; Davidson and Sauer, 1994) have shown that secondary and tertiary structures do appear frequently and spontaneously in random strings built upon suitably small alphabets. Many of the basic folding patterns of natural proteins can even be explained theoretically by assuming the randomness of their primary sequence (White, 1994): this suggests that the main carrier of folding information is the composition of amino acids rather than their linear ordering (Rahman and Rackovsky, 1995). All these clues, that nicely fit into the neutral theory of evolution, have oriented biochemists towards seeing modern proteins as memorized ancestral random polypeptides, that have been slightly edited by selection to optimize their active sites and stability under specific physiological conditions.
1
As Jaques Monod has put it (Monod, 1972):
In 1952, F. Sanger described the first complete sequence of a globular protein. This turned out to be both a revelation and a deception. This sequence, defining the structure and therefore the elective properties of a functional protein (insulin), did not show any regularity, characteristic feature, or limit. In those days it was hoped that, with the addition of further documentation, it might be possible to find the general laws of association and some functional correlations. Today we know hundreds of sequences corresponding to the proteins extracted from many different organisms. From them and their systematic comparison, performed with the help of up-to-date analysis and calculation devices, we can now deduce the general law: the chance law. More precisely, these structures are “random” because by knowing precisely the order of 199 residues of a protein containing two hundred it is not possible to formulate a theoretical or empirical law which allows us to predict the nature of the only residue still to be analytically identified.
Along with this intrinsic, evolutionary randomness, two additional problems make the definition of information in polypeptides even more elusive. The first problem is context: the translation of an aminoacid string into a three-dimensional structure is made possible by the cooperation of many distant substrings of the same and of different molecules (e.g., chaperones, multimers); the transport of many proteins to their proper cellular compartment and the acquisition of their final function depend on multiple post-translational modifications. Therefore, the information that leads a protein to assume its specific biological role is distributed in a context of interactions that trascends the single sequence (Adami and Cerf, 2000; Szpankowski and Konorski, 2007; Galas et al., 2008). The second problem is resolution: the key functions of a protein are often implemented by few atoms configured in specific spatial arrangements and bearing specific chemical properties. A single letter of the primary sequence of a protein hides tens of such atoms, positions and properties: these sub-symbolic signals are doomed to evade any measure of information that treats proteins as strings on the traditional amino acid alphabet.
Notwithstanding these fundamental issues, the question of what and how much information is carried by amino acid sequences has historically attracted a lot of attention, both for obvious purposes of classification, prediction and insight into folding and evolution, and for the screening and synthesis of artificial polypeptides for their use in new drugs (Davidson et al., 1995; Doi et al., 2005). Some successes have been recorded, especially in the context of large sets of non-homologous proteins—for example, the proteome of an organism (Macchiato et al., 1985; Adjeroh and Nan, 2006; Benedetto et al., 2007). Estimates of differential entropy and context-free grammar complexity (Weiss et al., 2000) have shown that the complexity of such large sets is lower than the complexity of a corresponding set of random strings by approximately 1%, about one third of which is caused by well-known low-complexity regions. Evidence of weak correlations at short, medium, and long range has also been found: positive correlations appear at medium range (≥100) and decrease with distance, implying that the amino acid distribution of proteins that are close in the genome is more similar than that of proteins far in the genome. The sign of the correlation between pairs of amino acids at medium distance forms groups that resemble those traced by widely accepted physico-chemical properties. Family-dependent, short-range periodicities in hydrophobicity, α-helix propensity, and charge have also been detected (Weiss and Herzel, 1998), and have been attributed to interactions between elements of the same secondary structures.
Both in statistical and in algorithmic information theory, the search for correlations and patterns is intimately related to the construction of compact models. Since a provocative 1999 study that advocated the incompressibility of proteomes (Nevill-Manning and Witten, 1999), there has been a modest flourishing of compression techniques tuned for long concatenations of polypeptides, spanning both the substitutional and the statistical realms (Giancarlo et al., 2009). We mention, among others, techniques consisting of instantiating the PPM algorithm with contexts of multiple lengths, weighted by amino acid mutation probabilities (Nevill-Manning and Witten, 1999); searching for exact and approximate reverse complements, repeats, and weighted context trees (Matsumoto et al., 2000); partitioning amino acids according to their frequency and invoking popular text compressors (Sampath, 2003); using amino acid substitution matrices to guide the creation of Huffman codes (Hategan and Tabus, 2004); building an off-line dictionary of variable-gap subsequences, constrained to be maximal in density and extension and to occur sufficiently frequently in the dataset (Apostolico et al., 2006); using panels of weighted experts that estimate the probability of a symbol using Markov models encoding species information, local context information; and repeated and complementary reversed substrings (Cao et al., 2007). These methods achieve entropies that range from about 3.67 to 4.05 bits per symbol, while other estimations based on the k-th order Shannon formula and Zipf curves reach 2.5 bps; incorporating secondary structure information in a gambling algorithm à la Shannon lowers this bound to about 2 bps (Strait and Dewey, 1996).
As expected, the analysis of stand-alone sequences has yelded more limited results. Measures of entropy over sliding windows have been shown to separate globular and fibrous proteins (Romero et al., 1999), and Lempel-Ziv complexity has been used to predict the cellular location of proteins (Xiao et al., 2005). Adding physico-chemical information to amino acids has enabled a Fourier analysis to detect characteristic periodicities in two protein families with similar structural architectures (Rackovsky, 1998); a mapping of recoded protein sequences onto one-dimensional Brownian bridges has revealed systematic deviations from randomness that have been related to energy minimization (Pande et al., 1994). The entropy of the primary sequence has also been shown to correlate with the inverse packing density and the hydrophobicity of residues in their spatial conformations (Liao et al., 2005).
In the present article, rather than identifying information with negentropy or compressibility, we correlate it to laws that govern the abundance of suitable combinatorial substructures of polypeptide strings. Rather than focusing on windows of fixed length or on substrings, we measure the composition of subsequences of any length, but constrained to occur with a predefined maximum gap between consecutive symbols, and to greedily choose the leftmost occurrence of each character. Subsequences can grasp long-range correlations in strings and, at short range, are well known to encode signatures and motifs that characterize protein families (Hulo et al., 2008). Rather than examining concatenations of many non-homologous polypeptides, we measure and compare the composition of each amino acid string in isolation. In addition to using our measures for comparing and classifying, we explicitly investigate their role in separating natural molecules from random permutations. This is perhaps the first time in which the vocabulary of all distinct subsequences of a set of structurally and functionally diverse polypeptides is systematically counted and analyzed.
The article is organized as follows: Section 2 formalizes the notion of constrained subsequence and of class of positional equivalence. In the spirit of Colosimo and De Luca (2000), Section 3 characterizes a set of sequences as extremal, in the sense that they cannot be enriched with characters without losing some occurrence in the string. Sequences and equivalence classes are then embedded in a natural spatial representation, in which they assume the form of paths and points, respectively. Section 4 describes suitable measures on this representation, and defines the set of polypeptides that have been selected for our experiments. Measures on subsequences are used for classification in Section 5, while Section 6 studies an array of laws that, in our dataset, are seen to relate these measures to string length and to the hiatus of subsequences in our dataset. Finally, Section 7 analyzes regularities that constrain pairs of measures in random permutations of our dataset.
2. Preliminaries
Given a string s of characters from an alphabet Σ, a subsequence of s is any string v that can be obtained by removing from s one or more, not necessarily consecutive characters. An occurrence of v in s is specified by a list of positions of s matching the characters of v consecutively. The positions of s that correspond to the first (respectively, last) character of v form the left (respectively, right) list of v, denoted by
Given an ω-occurrence
If ω = 1, each horizon contains only one window and the panorama cannot contain more than |Σ| + 1 windows in total. To examine a more elaborate case, let
Word v has ω-occurrences:
Note that the number of ω-occurrences of subsequences of length k occurring at the same starting position in s is
Definition 1 (Left equivalence)
Two subsequences v and w are left equivalent, denoted v ≡ l w, if
We stipulate that strings never occurring in s are assigned to the class characterized by the empty list. We also assign the left list
Property 1
If v ≡ r w, then
Property 2
The relation ≡
r
is right-invariant, i.e., v ≡ r w implies
Note that v and w can have the same panorama even though they do not have the same number of occurrences in s, and that
Definition 2 (Implication)
We say that w implicates or induces v on s if for every occurrence
Clearly, implication is not symmetric, e.g., with s = ACAGTTT$$$, v = AGT and w = ACT, we have that w implicates v even though v does not implicate w.
Definition 3 (Equivalence)
Two subsequences v and w of s are equivalent, denoted v ≡ w, if they implicate one another.
We say that a class of the equivalence relation ≡ is a terminal class if the list of its right occurrences is {|s|}. Every subsequence in such class is called terminal subsequence.
Lemma 1
If v ≡ w, then
Lemma 2
The equivalence relation ≡ is right-invariant.
Note that
3. Special Subsequences and The ω-Suffix Space
It is of interest to single out the subsequences of s that cannot be expanded without losing support, i.e., their number of ω-occurrences in s. The following definition may be considered an extension to subsequences of the one applied to substrings in Colosimo and De Luca (2000).
Definition 4 (Special subsequence)
A string
Note that, according to this definition, ɛ is a special subsequence. Special subsequences have the following properties.
Property 3
Let
Property 4
If av is a special subsequence and
Property 5
If v is a special subsequence, then such is also any w ≡ v.
Following is the dual notion of that of a special sequence.
Definition 5 (Antispecial subsequence)
A subsequence v of s is antispecial if any extension va of v in s,
Therefore, a subsequence is antispecial if and only if every symbol with which it can be extended in s appears in every horizon. Notice that a subsequence v that is extensible in s in only one way, or such that
The definition of special sequence embodies a criterion to build all the ≡
l
and ≡ classes of a string s. Assuming that all the occurrences
Like standard common subsequences, also those considered here are susceptible to a natural geometric representation. Let
A greedy ω-subsequence corresponds to a chain in this partial order such that, for each pair of consecutive points
The simple construction that we now proceed to describe traces all the sequences that ω-occur greedily in s, resulting in what constitutes an expansion of the constrained Hasse diagram above. The space Ψ
a
(s) sets the natural stage also for such a construction, which starts at point
Let V be the set of the points in space Ψ a (s) that are visited by the algorithm just described, and let A be the set of the arcs, oriented and labeled by symbols of Σ, that indicate the extensions of each point of V carried out by the algorithm. Graph Ga(s) = (V, A) is called the ω-suffix graph induced by symbol a on s.
Lemma 3
The points of Ga(s) represent all and only the classes of the equivalence relation ≡ among the sequences starting by a that ω-occur greedily in s.
Space Ψ a (s) may have a very high number of dimensions, but it contains subspaces of smaller dimensionality:
Definition 6 (Subspace of Ψ a (s))
Let
Actually, strings belonging to the same class under ≡ l are projected to paths of Ga(s) ending at points located in the same subspace of Ψ a (s).
In particular, the class
4. The Dataset and Its Rationale
In the following sections, we set up a battery of experiments to study the properties of the suffix graphs generated by amino acid strings and by their random permutations. Natural compositional measures that we will consider are the number of points (special, antispecial, normal, terminal), the number of arcs (internal and external) and the number of subsequences (special, antispecial, normal, terminal) at different values of ω. The following sections will aggregate and systematize over one million data points.
Many previous investigations (Nakai et al., 1988; Weiss and Herzel, 1998; Weiss et al., 2000; Lapinsh et al., 2002; Li et al., 2003b) have recoded the original amino acid strings with reduced alphabets that incorporate physico-chemical scales. The large number of scales published (Kawashima et al., 2008), and the lack of a standard methodology to perform such recoding haunted our preliminary experiments with many parameters that made the analysis dependent on the fine-tuning of the scale quantization. Amino acid similarities are also known not to be universal: at different positions of a protein, different sets of amino acids or of amino acid substrings can more likely substitute for one another (Han and Baker, 1995), making a fixed substitution scheme less biochemically significant. The necessity to make our results as general as possible led us to analyze polypeptides encoded in the original alphabet of amino acids.
Most proteins consist of modular subunits (called domains) whose spatial conformation and function are thought to be independent of other parts of the protein. A limited number of highly similar domains are seen to occur in all known proteins, both as parts of larger multidomain structures and by themselves (Richardson, 1981; Murzin et al., 1995; Orengo et al., 1997), suggesting that they are remnants of ancient functional polypeptides that have been assembled by evolution to produce the combinatorial variety of structures and functions that appear in modern proteins. The modular nature of domains makes them better candidates than whole proteins for investigating regularities and patterns, since the concatenation of different domains could be a source of noise. The sequential compactness and the moderate length of domains make them preferable to secondary structures, supersecondary structures, or motifs, that are typically shorter and non-contiguous subsequences.
The SCOP (Structural Classification of Protein) database (Murzin et al., 1995) is a comprehensive ordering of all protein domains of known structure according to their evolutionary, structural and functional similarity. The basic classification unit is the domain, that is put at the leaves of a tree having three more hierarchical levels: families, containing domains with a common evolutionary origin as testified by high sequence similarity or highly similar function and structure; superfamilies, containing domains with low sequence similiarity but sharing structural and functional features that suggest a common evolutionary origin; folds, containing domains with a specific set of major secondary structures, a specific configuration of these structures in space, and a specific connection pattern; and classes, containing domains that share the same frequency of secondary structures (e.g., domains in which the large majority of secondary structures are α-helices). This classification is manually curated by biologists.
We elect a subset of 148 domains of SCOP as our main dataset: we will refer to this dataset as D1 in what follows (Table 1). We choose domains to span two different classes (1 and 2), two different folds per class (1.1, 1.8, and 2.1, 2.2), two different superfamilies per fold (1.1.1, 1.1.2, 1.8.1, 1.8.4, and 2.1.1, 2.1.2, 2.2.2, 2.2.3), and two different families per superfamily 2 (1.1.1.3, 1.1.1.4, 1.1.2.1, 1.1.2.2, 1.8.1.1, 1.8.1.2, 1.8.4.5, 1.8.4.6, and 2.1.1.1, 2.1.1.2, 2.1.2.1, 2.1.2.2, 2.2.3.2, 2.2.3.3, 2.2.2.1, 2.2.2.2). The purpose of this selection is twofold: on one hand, we want to determine at which level of accuracy different measures on suffix graphs reconstruct the SCOP classification. For example, if a measure correctly separates domains in different classes but not in different folds, we can conjecture that the measure grasps information encoded in the dominant secondary structures and not in their spatial arrangement. On the other hand, we want to test whether the primary sequence of domains systematically differs from random strings, and if so whether this difference is a widespread phenomenon or it is confined to specific leaves of the classification. To do so, we analyze 100 random permutations of each string in D1.
As shown in Figure 1, all domains use 15–20 symbols, and have empirical entropy 3 of 3.5–4.2; however, entropy in the same SCOP leaf can vary widely inside this range. Conversely, string length and the compression ratio achieved by a popular string compressor, are uniform inside many leaves (notably 1.1.1.3, 1.1.1.4, 1.1.2.1, 1.8.4.6, 2.1.1.1, 2.1.1.2, 2.1.2.1, 2.1.2.2, 2.2.3.3, 2.2.2.2), and they display trends that are very similar to each other. Not surprisingly, a significant proportion of domains are either expanded or not compressed.
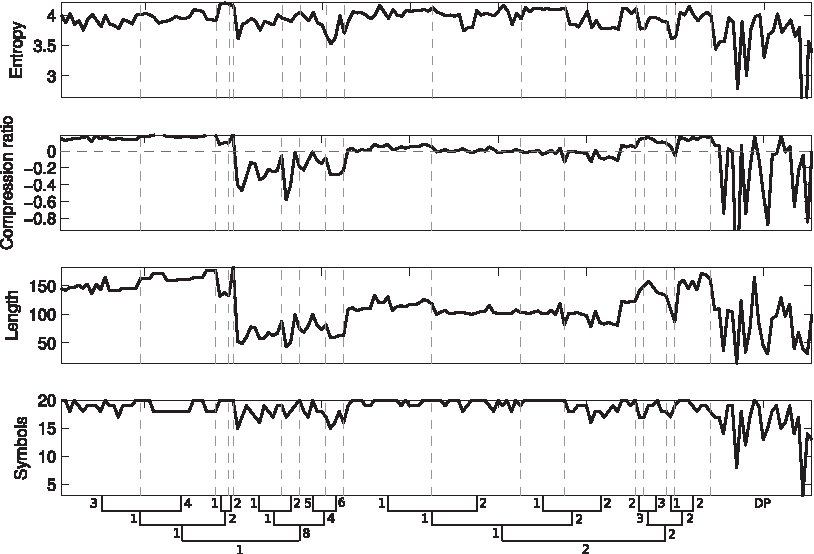
Measures on strings in D1 ∪ D2. Empirical entropy is computed using logarithms to base 2, where 0 · log 2(0) is set to 0. Compression ratio is defined as (|s|-|s′|)/|s|, where s is the original string, s′ is its compression with gzip –best, and | · | is the size in bytes.
Modern proteins do not consist entirely of domains: some regions have no fixed spatial configuration under physiological conditions, but are capable of dynamically transitioning through an ensemble of structures (Richardson, 1981; Wright and Dyson, 1999; Sickmeier et al., 2007). The flexibility of these unstructured (or disordered) segments allows them to fold and bind to a target simultaneously, transitioning from disorder to order according to their biochemical environment: this allows a single protein to bind multiple targets, and different proteins to bind the same target, an important feature in signalling and regulation networks. The fluctuation of spatial conformation is also exploited to create regions of exclusion in space, to facilitate phosphorylation and acetylation, and to capture small molecules. In disordered regions, the relationship between sequence and structure is different than in typical folded domains: disordered regions are known to be enriched in charged and polar, and depleted in hydrophobic residues. Along with other chemical and spatial indicators, these biases have been used to construct various disorder prediction heuristics (Li et al., 2000) and to classify disordered regions into subclasses. From the purely syntactic point of view, disordered regions tend to have low entropy (Romero et al., 2000; Weathers et al., 2006), however some disordered sequences have high entropy and some low-entropy sequences are not disordered.
DisProt (Sickmeier et al., 2007) is a comprehensive functional classification of all polypeptide regions for which there is experimental evidence of disorder. We collect a subset of 23 regions of DisProt in a secondary dataset (called D2 in what follows; Table 2); the choice of strings in this set is again arbitrary, except that for efficiency and consistency we consider only proteins having a single disordered region of length at most 200. The purpose of this dataset is twofold: on one hand, we want to test whether disordered regions differ from domains according to measures on suffix graphs. We conjecture that if a measure clearly separates D1 from D2, then it grasps information that only polypeptides with a fixed spatial conformation encode. On the other hand, we want to test whether disordered regions can be distinguished from random strings, and whether such difference resembles those that intercur between domains and random strings. To do so, we analyze again 100 random permutations of each string in D2.
As shown in Figure 1, just 8 regions in D2 use less than 15 symbols, and just 5 regions have empirical entropy less than 3.5, the minima in D1. Furthermore, just 8 regions have positive compression ratio, and compression ratios in D2 are never larger than the largest compression ratio achieved in D1. Therefore, strings in D2 do not appear as systematically “less complex” than strings in D1. Six disordered regions have compression ratio smaller than − 0.58, the minimum in D1, but this does not allow to conclude that disordered regions are systematically “more complex” than strings in D1, either.
5. Classifying with Suffix Graphs
Figure 1 shows that entropy and number of symbols cannot be used to group the dataset into SCOP regions, but that compression ratio and length are largely constant inside some SCOP leaves, and that values in different leaves can be clustered around approximately three levels: “high” (for folds 1.1 and 2.2), “medium” (for 2.1), and “low” (for 1.8). We want to test whether any measure on suffix graphs achieves a similar grouping, and whether these groups have a parallel in the classification of SCOP.
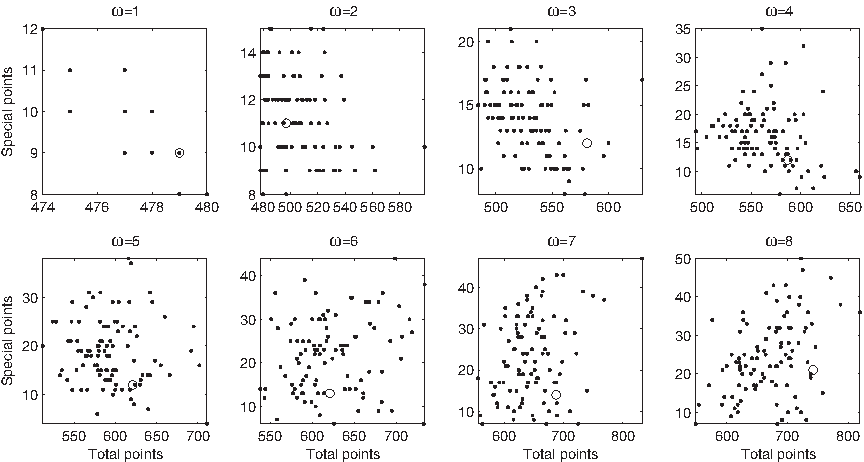
No single measure considered in its absolute value displays such a clustered behavior, 4 however groups of similar values seem to appear when measures are taken relative to the total number of their respective elements: for example, the number of special points divided by the total number of points (Fig. 2) assumes approximately three distinct values at every ω < 3: “low” (for folds 1.1 and 2.2), “medium” (for 2.1), and “high” (for 1.8); D2 does not form a group of its own: some disordered regions assume values that are significantly larger than domains, while others fall in the range of D1. A similar pattern occurs in antispecial points, internal arcs and external arcs at ω < 3; the uniformity inside SCOP leaves, on the other hand, decreases in all measures when increasing ω beyond 3. Terminal points are an exception: both their clustered trend and their values remain approximately the same up to ω = 8.

Number of special points divided by the total number of points across D1 (bottom) and D1 ∪ D2 (top) at ω = 2. Similar trends appear at ω = 1, 3.
This consistency across different measures suggests that the relative abundance of points and arcs could be the foundation of a coherent clustering criterion. Representing each string as a vector in the four-dimensional simplex of the relative number of special, antispecial, normal and terminal points, and setting ω = 1, 2, 3, 4, some disordered regions (respectively 8, 7, 9, and 7 out of the 23 of D2) are separated from the rest of D1 ∪ D2 into an independent subtree, accompanied by some members of fold 1.8 (Fig. 3). This group of strong outliers is partitioned, in its turn, into two or three well-separated subgroups at all ω ≤ 4. The following strings are outliers at ω ≤ 4:
DisProt 34: GGGSGGGSGGGSEGGGSEGGGSEGGGSEGGGSGGGSGSG, part of attachment protein G3P; DisProt 9: DSHRDASQNGSGDSQ, part of transcription initiation factor IIA small subunit; DisProt 13: DPRFQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ, part of choriogonadotropin subunit beta;
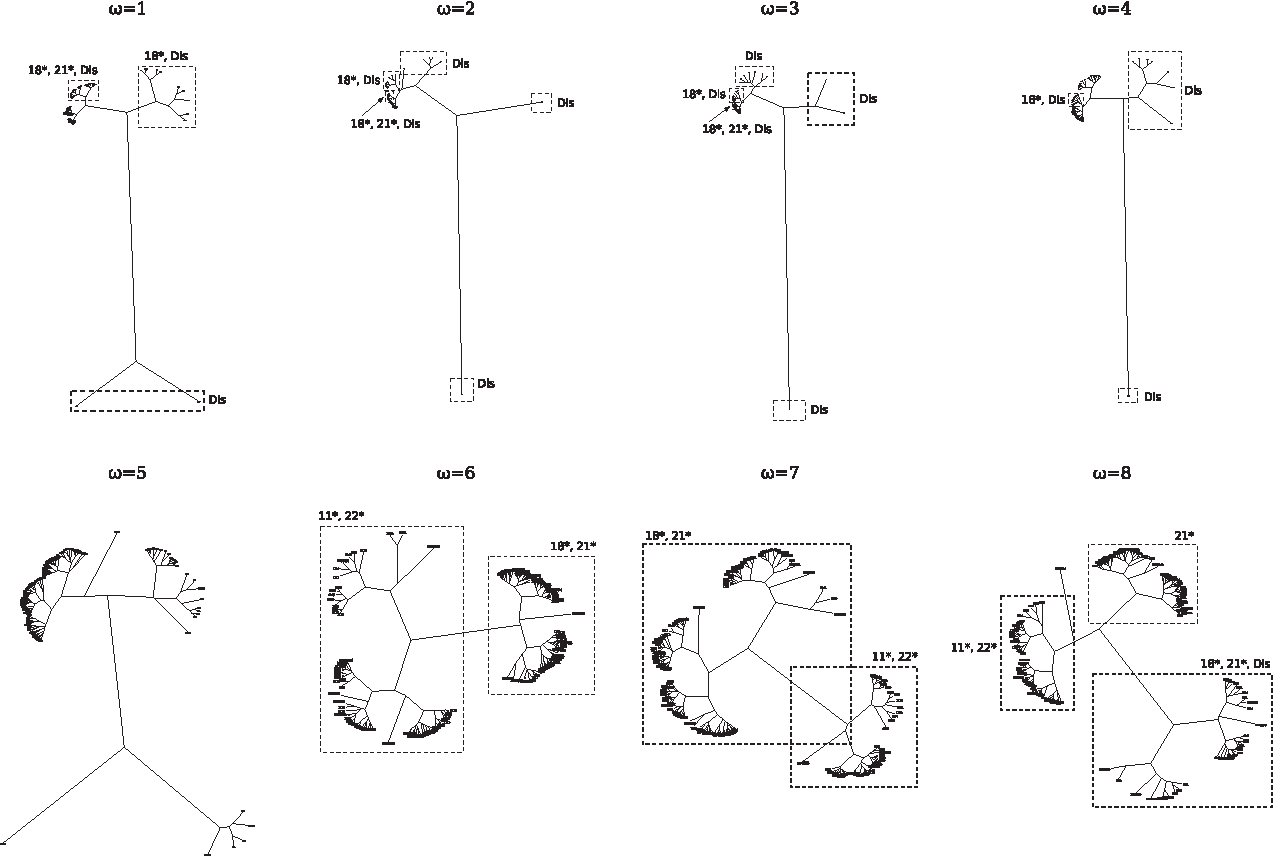
UPGMA trees between four-dimensional vectors that collect the relative number of special, antispecial, normal, and terminal points of each string in D1 ∪ D2. Distances are Euclidean. Branch lengths are drawn proportional to distance. Disordered regions are denoted with “Dis.”
and the following additional strings are outliers at ω ≤ 3:
DisProt 35: GCTLSAEDKAAVERSKMIDRNLREDGEKAAR, part of guanine nucleotide-binding protein G(i), alpha-1 subunit; DisProt 4: LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES, part of cathelicidin antimicrobial peptide.
These strings are among the shortest in D2, but only DisProt 34 stands out for its apparent regularity, and is put farthest from the rest of the dataset in the trees. At ω ≤ 3 most strings in fold 1.8, as well, are strongly separated from the rest of D1: these strings form two subtrees, one containing some strings in D2 and the other containing some strings in D2 and 2.1. A closer look at the densities at ω < 4 (Fig. 5) shows that the average number of special, normal and terminal points in D2 is at least 2, 4, and 2 times larger than the respective values in D1, respectively, and that the average number of terminal points in fold 1.8 is approximately two times larger than the average number of terminal points in the rest of D1. Strong outliers in D2 have an average number of special, normal and terminal points that is at least 3, 5, and 2 times the rest of D2, respectively, while special and normal points do not separate 1.8 from D1 at any ω > 2.
At ω = 5, the shape of the tree seems to experience a phase transition: there are still at least three sharp partitions with clear subpartitions, but none of these subtrees either consists predominantly of disordered regions, or reflects a SCOP class. At ω = 6, another transition seems to occur: the tree becomes separated into two groups associated with folds 1.1–2.2 and 2.1–1.8, respectively, but the interior of each group does not further reflect the SCOP hierarchy or the division between D1 and D2. At ω = 8 the tree becomes organized into 3 groups: one containing members of D2, 1.8 and 2.1, another one containing strings in 2.1, and the third containing strings in 1.1–2.2. DisProt 34, 9 and 13 are put consistently far from the other groups at all ω ≥ 5, but they never form an independent subtree.
By discarding branch length information, more details about the topology of the trees can be appreciated (Fig. 4). At ω = 1, three distinct subtrees 5 for folds 1.1, 2.1, and for a mixture of 1.8 and D2 can be identified, as well as subtrees for families 2.1.1.1, 2.1.1.2, 1.1.1.3, and 1.1.1.4. Most members of fold 2.2 lie in the subtree of fold 1.1, while the disordered regions that are not strong outliers are dispersed mainly in the 2.1 and in the 1.8-D2 subtree. Setting ω = 2 shows a similar structure, but in this case the 1.1 subtree contains a large subtree of 2.1 strings, and families 2.1.1.1 and 2.1.1.2 do not form compact trees. Fold 2.2 still tends to merge with fold 1.1. At ω = 3 only the subtree of strings in 1.8 and D2 survives, while the rest of the tree does not resemble SCOP any more. At ω = 4, 5 no subtree keeps a strong resemblance to a SCOP group. Finally, at ω ≥ 6, no additional structure can be detected inside the subtrees marked in Figure 3.
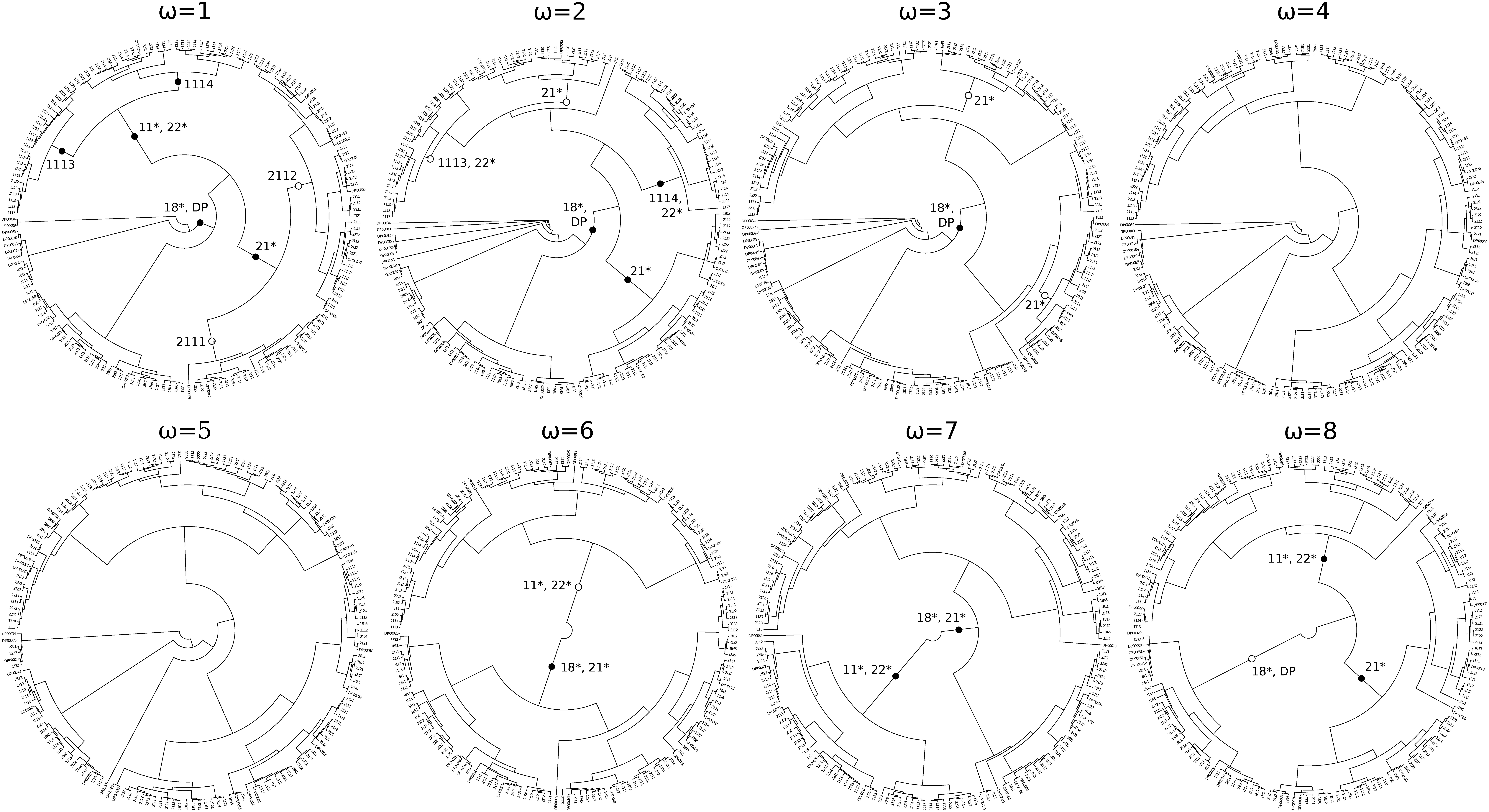
The trees of Figure 3 with branch lengths not drawn proportional to distance. Each string is labeled by the identifier of its SCOP family; disordered regions are labeled by their DisProt ID. White dots mark subtrees for which a SCOP group exists whose Jaccard coefficient with the subtree is at least 0.3; black dots mark subtrees for which a SCOP group exists whose Jaccard coefficient with the subtree is at least 0.5. The identifiers of the SCOP groups that have maximum Jaccard coefficient with each subtree are noted near the dots.

Relative proportion of special points (black, bottom), antispecial points (light gray), normal points (black, top), terminal points (white) at ω ≤ 4. On the horizontal axis, from left to right (separated by dashed lines): the subset of strong outliers of D2, the rest of D2, fold 1.8, and the rest of D1. For clarity of presentation, not all members of D1 are shown.
The topological phase transition that takes place at ω = 6 can be assessed quantitatively using the approach proposed in Ferragina et al. (2007). Given a threshold distance τ, we treat a distance matrix M as a binary classifier that puts strings i and j in the same group iff M(i, j) < τ. Using a specific level of the SCOP tree as reference, we measure the false positive (FP) and the true positive (TP) rate of this classifier for different choices of τ: plotting these values on the FP/TP space traces the receiver operating characteristic (ROC) of the classifier. The characteristic of a classifier that perfectly reproduces the SCOP classification would be locus {FP = 0} ∪ {TP = 1}, while the ROC of a random guess would be {FP = TP}; in general, the larger the area under the curve, the closer to SCOP is the classificator. The ROC analysis for folds, superfamilies and families (Fig. 6) shows that adherence to SCOP decreases when ω is increased from 1 to 5, but then increases when ω is set to 6, 7, and 8. At the class level, the increase of classification ability at high ω is less evident. Classification performance is approximately the same at the fold, superfamily and family level.
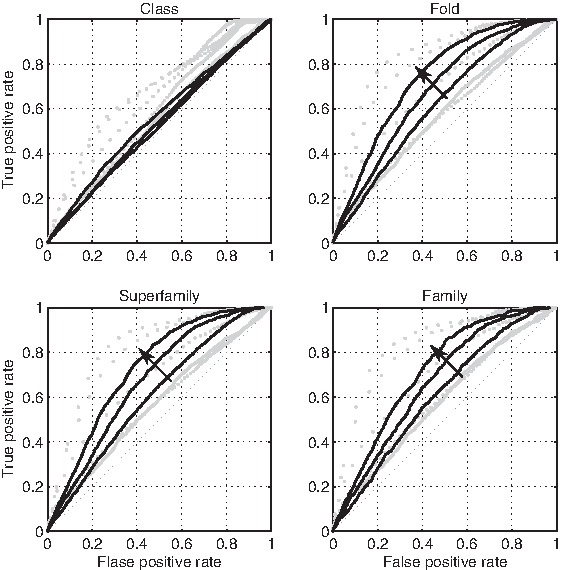
The relative composition of points used as a classifier of SCOP levels. Gray dotted lines are ROC curves for ω ≤ 5; black lines are ROC curves for ω ≥ 6, with ω increasing in the verse of the arrow. D2 is considered only at the class level.
Describing the trees produced by different choices of the tree-construction algorithm, as well as fine-tuning the distance and the combination of measures to achieve the best resemblance to SCOP, is beyond the scope of this paper. Here we just note that arcs give results similar to points, but the relative proportion of special, antispecial, normal and terminal subsequences is not capable of partitioning D1 into SCOP groups for any ω > 1 (Fig. 7): few strings are put very far from the rest of the dataset (among which DisProt 34 and DisProt 9), but this group of outliers does not correspond to any SCOP cluster. Other well-separated subtrees do appear in this classification, but they have no clear SCOP analogue.

The relative composition of subsequences (ω > 1) used as a classifier of SCOP levels. D2 is considered only at the class level.
We remark that the ability of suffix graph measures to detect biologically meaningful groups of polypeptides is surprising, because the distance between two strings is computed by comparing their compositions, and not by counting patterns or substructures that occur in both. Recall also that strings have not been recoded with alphabets that incorporate biochemical information. It is therefore natural to ask how these trees compare to those derived from techniques that do exploit common features and biochemical properties: we expect the latter to strongly separate all strings in D2 from D1, and to reproduce the shape of SCOP more accurately.
Current measures of dissimilarity between biological strings can be grouped into essentially two families. Alignment-based measures (Apostolico and Giancarlo, 1998; Sankoff and Kruskal, 1999), popularized by the BLAST package, define similarity as a function of the local alignment score computed using the Smith-Waterman algorithm with a highly tuned penalty function. Even though alignment-based similarity notoriously suffers from methodological and technical limits when applied to whole genomes, it is ideally suited for searching proteins and short nucleotides in databases. We test the classification ability of alignment-based similarity by computing, for each pair of strings x,y in D1 ∪ D2, the distance d(x, y) = (1 − b(x, y)/b(x, x)) · (1 − b(y, x)/b(y, y)), where b(x, y) is the bit score of the alignment between x and y computed by blastp, a version of BLAST specifically designed for protein-protein comparison and using the BLOSUM62 amino acid similarity matrix.
Alignment-free techniques (Vinga and Almeida, 2003; Qi et al., 2004; Ulitsky et al., 2006; Apostolico and Denas, 2008; Sims et al., 2009; Apostolico et al., 2010) compare two strings using the frequency of common substructures (typically short substrings or subsequences), thereby implicitly projecting a string onto a vector that belongs to the space generated by the family of these substructures. The most ancient such technique projects a string into the space of all possible substrings of a fixed length k, and uses geometric or statistical measures to estimate the distance between two vectors. Since the effectiveness of these methods critically depends on k (Wu et al., 2005), it will not be examined here. Another notable subclass of alignment-free techniques resorts to mutual compressibility: the Normalized Compression Distance (NCD) (Cilibrasi and Vitányi, 2005; Kocsor et al., 2006; Ferragina et al., 2007), for example, approximates the Normalized Information Distance, a non-computable measure of dissimilarity hinged on Kolmogorov complexity that has been shown to be a lower bound to every possible distance measure between two strings (Li et al., 2003a). NCD summons a textual compressor on the concatenation of a pair of strings, thereby exploiting the features occurring in both that the compressor is designed to detect.
We test the classification ability of alignment-free distances by using NCD with the general-purpose Lempel-Ziv compressor gzip. A special purpose, highly-tuned protein compressor could approximate better the ideal compressor that should be used with NCD. Unfortunately, none of the protein compressors listed in Section 1 is both publicly available and mature enough for usage. Since many of them resort to exact and approximate reverse complements, we experiment with the popular gencompress (Chen et al., 2000), based on similar principles, even though we are aware that this program was designed for DNA compression.
The classification performance of the composition of points at ω ≤ 3 turns out to be comparable or superior to those of blastp, gzip and gencompress at the fold and superfamily levels (Fig. 8). At the family level, the composition of points has a performance comparable or superior to both gzip and gencompress, but inferior to blastp, while at the class level the composition of points is inferior to gzip and gencompress, but comparable to blastp. At ω ≥ 7 (Fig. 9) the composition of points is comparable to gzip and gencompress at all levels except class, but is inferior to blastp at all levels except fold and superfamily.
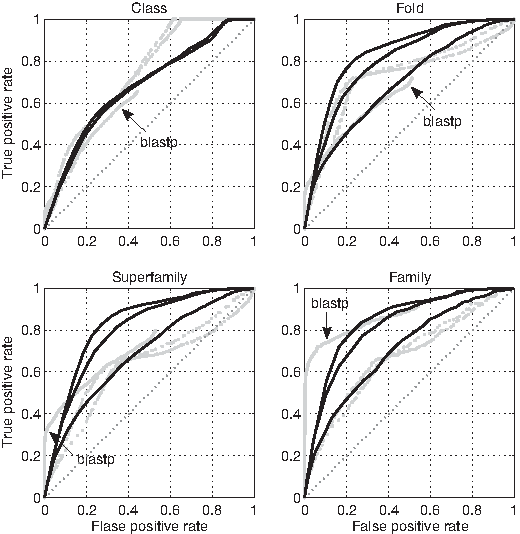
ROC curves for blastp (with default parameters), gzip –best and gencompress distances (gray dotted lines), compared to the ROC curves for the relative number of points at ω ≤ 3 (black solid lines). The blastp curve is truncated because low-scoring alignments are not outputted by the program. D2 is considered only at the class level.

ROC curves for blastp (with default parameters), gzip –best and gencompress distances (gray dotted lines), compared to the ROC curves for the relative number of points at ω ≥ 7 (black solid lines). The blastp curve is truncated because low-scoring alignments are not outputted by the program. D2 is considered only at the class level.
UPGMA trees built with both alignment-free distances sharply separate D1 from D2 (Fig. 10), while the blastp tree is not capable of grouping all members of D2 under a single subtree. Both alignment-based and alignment-free trees contain macroscopic differences with respect to the SCOP hierarchy, putting a class or fold in the subtree associated with another. Furthermore, drawing branch lengths proportional to distance produces alignment-free trees that are largely devoid of structure (Fig. 10): most strings in D1 are put at approximately the same distance from all other strings in D1. In comparison, the blastp tree is more structured, but longer branches do not correspond to major SCOP divisions. Finally, no strong outlier can be detected in any tree.
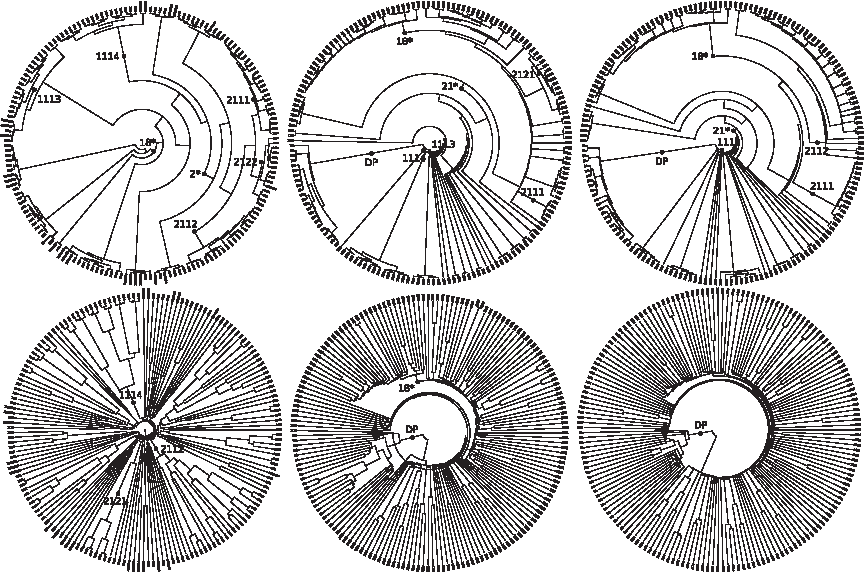
UPGMA trees induced by (from left to right): blastp scores (with default parameters), the Normalized Compression Distance with compressor gzip –best, and the Normalized Compression Distance with compressor gencompress. First row: branches not drawn proportional to distance. Second row: branches drawn proportional to distance. NCD(x, y) = (min{C(xy), C(yx)} − min{C(x), C(y)})/max{C(x), C(y)}, where C is a normal string compressor.
6. Laws Governing Polypeptides
As stated in the introduction, our aim is to identify laws constraining the suffix graphs of polypeptides. In this section, we investigate the dependence of suffix graph measures on string length and on the the hiatus of subsequences.
6.1. Dependence on string length
The comparison of Figures 1 and 2 suggests that the number of special points taken relative to the total number of points is inversely proportional to string length. At ω = 1 this inverse proportionality comes not unexpected: the total number of points (in this case, distinct substrings) grows at most as the square of string length, and the number of special points (in this case, equivalent to the number of distinct special substrings) grows at most linearly with string length; therefore, the ratio (Special points/Total points) should behave like a/n + b, where n is string length and a, b are suitable constants, assuming the strings in the dataset to be approximately random. In principle, every string in D1 ∪ D2 could obey to a different set of parameters, making D1 ∪ D2 appear as a disordered cloud in the space generated by the (Special points/Total points) ratio and string length. However, in light of the homogeneities of Figures 1 and 2, and of the trees built in the previous section, we expect to see a limited number of distinct curves along which domains in similar SCOP groups are aligned. These curves (that we will also call loci) should be signatures of such groups, and their detection could guide classification.
Plotting the relative number of special points versus string length at ω = 1 (Fig. 11) shows indeed the expected 1/n proportionality, but surprisingly the majority of strings in D1 ∪ D2 are aligned along the same locus with coefficients 6 a ≈ 1.435, b ≈ 0. Significantly, the only strong outlier is DisProt 34. There are three features that make DisProt 34 unique in the dataset: its highly repetitive structure, the use of just 3 distinct symbols, and its small entropy ( ≈ 1.215). The locus could therefore reflect a property of all strings that lack a strong periodic structure, that have a sufficiently high number of symbols, a sufficiently high entropy, or any combination of these three features. To test this hypothesis, we collect an additional dataset consisting of 89 distinct SCOP domains of length at most 30: we will refer to this set as D3 in what follows. It turns out that D3 contains at least two strings (tumor necrosis factor receptor superfamily member 17, BCMA, csqneyfdsllhacipcqlrc; nucleic acid binding protein p14, kgpvcfscgktghikrdckee) that lack a strong periodic structure, use a number of symbols comparable to strings in D1 ∪ D2 (14 and 13, respectively), have entropy comparable to strings in D1 ∪ D2 ( ≈ 3.5398 and ≈ 3.8643, respectively), but that do not lie on the locus. This proves that the locus cannot be explained by any combination of the candidate quantities alone. We also note that low entropy alone does not force a string to lie outside the locus: Figure 12 shows that random strings on 20 symbols and minimum entropy ( ≈ 1.1169, even lower than DisProt 34) can lie on the curve.

Relative number of special points versus string length, in domains (dots) and disordered regions (circles). Strings in D3 are represented with gray crosses. The best interpolating a/n + b curve is shown as a black line.

The graph of Figure 11 at ω = 1. (
A locus with similar parameters persists at ω = 2, 3, with DisProt 34 and some strings in D3 continuing to be outliers. Increasing ω beyond 3 gradually transforms the sharp locus into a dispersed cloud that keeps no resemblance to the original curve. At ω ≥ 6 three disordered regions (DisProt 34, 13, and 19) become clearly separated from the rest of the dataset, along with few strings in D1.
We expect a direct proportionality between the number of special points y (not normalized) and string length n, and in particular a linear relationship y = an + b when ω = 1. Plotting these two quantities together (Fig. 13) shows indeed a linear bundle centered around a ≈ 0.317, b ≈ 0.318 for all domains and disordered regions except DisProt 34 and 25 (part of Fibronectin-binding protein A). The locus remains linear at ω = 2, but from ω = 3 it progressively tends towards a sparse nonlinear shape that we will call “horn.” We note that, at ω ≥ 6, this shape includes strings that were outliers at lower ω, in particular the highly regular DisProt 34. Strings in D3 are never seen to escape the locus, but random strings on 20 symbols with minimum entropy turn out to be outliers for every ω, proving that the curve is not an unavoidable regularity of all strings.

Number of special points (not normalized) versus string length, in domains (dots) and disordered regions (circles). Gray crosses are random strings on 20 symbols with minimum entropy.
The total number of points assumes the expected quadratic shape at ω = 1, which persists up to ω = 4, then it gradually becomes a horn (Fig. 14). Neither D3 nor DisProt 34 escape the locus, but few other disordered regions do. As before, it can be shown that there is at least one string that does not lie on the locus.
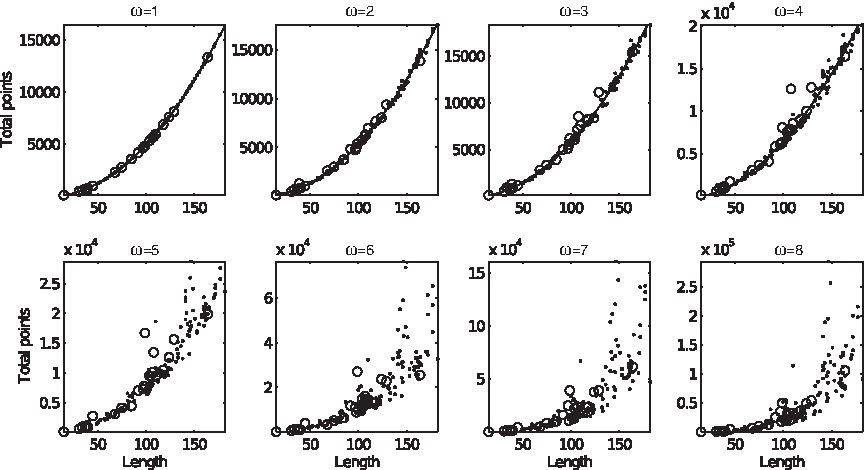
Total number of points versus string length, in domains (dots) and disordered regions (circles). The interpolating line is y = a*n2 + b*n + c. At ω = 1, a ≈ 0.4963. At ω = 2, a ≈ 0.5453. At ω = 3, a ≈ 0.5585. At ω = 4, a ≈ 0.6057.
Similar curves appear when other measures are considered: in all cases, at most one sharp curve appears, collecting the majority of D1 ∪ D2 with the possible exception of few outliers. Rather than analyzing each one of these curves in detail, we prefer to concentrate the attention on two measures in which the shape of the locus changes in a different way as a function of ω. In the relative number of antispecial points (Fig. 15) a sharp nonlinear curve of direct proportionality with few, strong outliers persists up to ω = 2, then it becomes disordered at ω = 3, 4, 5, and finally transitions towards a bundle of inverse proportionality at ω = 8. The second notable example is the relative number of normal points: no clear locus appears at ω ≤ 6, but a linear bundle starts to emerge at ω = 7, 8 (Fig. 16).
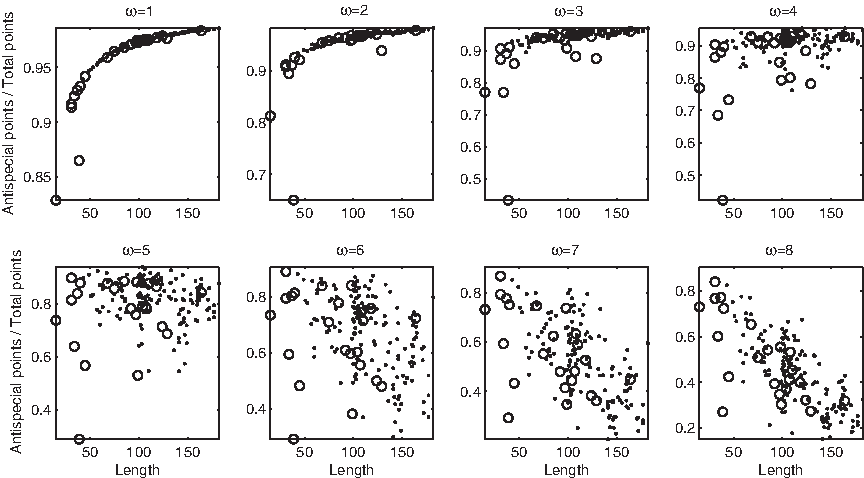
Relative number of antispecial points versus string length, in domains (dots) and disordered regions (circles). At ω = 2, 3, 4 some outliers fall outside the displayed range.
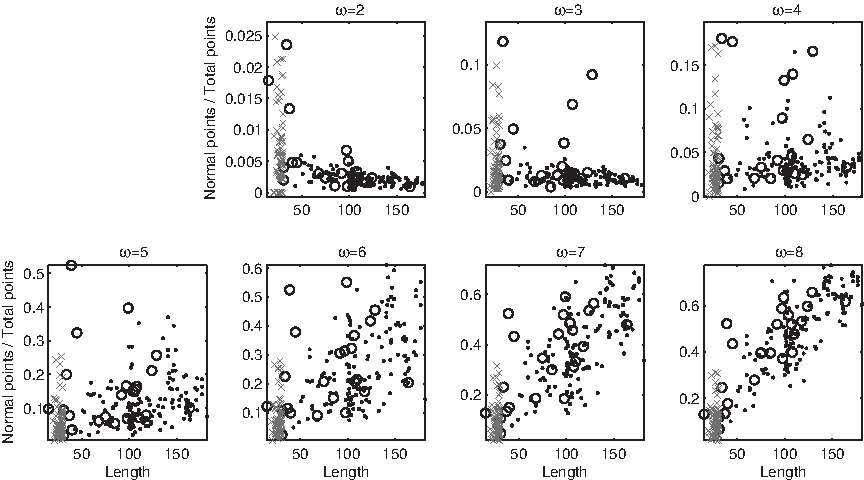
Relative number of normal points versus string length, in domains (dots) and disordered regions (circles). At ω = 2, 3 some outliers fall outside the displayed range.
These patterns of dependency of the relative number of special, antispecial and normal points on string length for specific values of ω largely explain the phase transitions seen in the trees of the previous section. At ω ≤ 3, the relative abundance of special and antispecial points is strictly controlled by string length, special and antispecial points are the most numerous types of point, and the tree reflects the three clusters induced by string length. At ω ≥ 7, antispecial and normal points are the most abundant type, their density is loosely influenced by length, and the trees show again a bi- or tripartition that reflects string length. When
Even though the trees of Figure 3 are largely a reflection of the groups induced by string length, we remark that this variable alone is not sufficient to explain some key features of the trees, like the strong separation of some members of D2 from D1 and of fold 1.8 from D1.
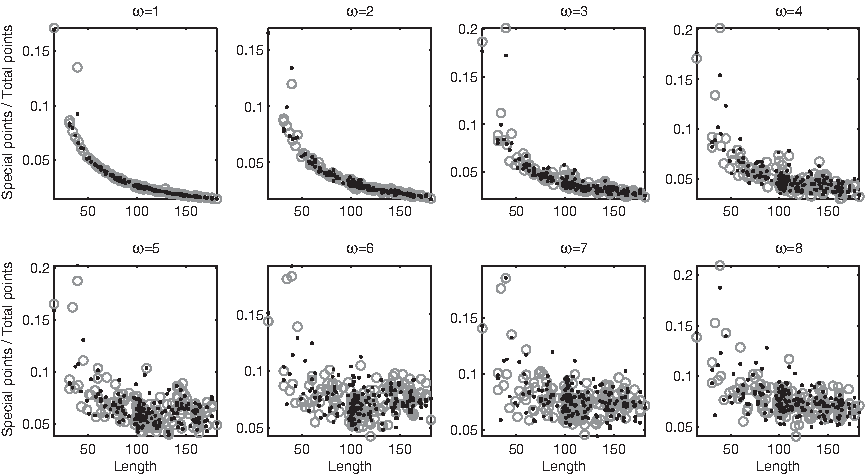
The trees described in the previous section showed that the information encoded by the relative abundance of subsequences differs from the information encoded by the relative abundance of points. Plotting measures on subsequences versus string length reveals that what these measures are partially lacking is the dependence on string length itself. For example, the number of antispecial subsequences (not normalized) grows exponentially with string length, and their growth is confined inside a bundle whose width expands with length (Fig. 17). However, there is no correlation between the relative number of antispecial subsequences and string length when ω > 1 (Fig. 18). On the other hand, the relative number of normal subsequences depends on string length under a relationship of exponential inverse proportionality for all ω ≤ 5 (Figure 19); this locus disappears into a disordered cloud at ω ≥ 6.

Number of antispecial subsequences versus string length, in domains (dots) and disordered regions (circles).

Relative number of antispecial subsequences versus string length, in domains (dots) and disordered regions (circles). At ω > 1 few strings lie far from the main cloud, and fall outside the displayed range.

Relative number of normal subsequences versus string length, in domains (dots) and disordered regions (circles).
A measure can have no dependence on string length at a specific ω, but it could nonetheless obey other rules. We have just seen that the relative number of normal points has no locus at ω < 7, and that the relative number of normal subsequences has no locus at ω ≥ 6: plotting these two measures one versus the other shows again no correlation for D1 ∪ D2, but it reveals that D3 is aligned along a horn at all values of ω > 1 (Fig. 20). No such regularity occurs, however, when we plot the relative number of antispecial points versus the relative number of antispecial subsequences.

Relative number of normal subsequences versus relative number of normal points, in D3 (black crosses), D1 (light gray dots), and D2 (light gray circles).
6.2. Dependence on ω
In the previous section, we have shown that the shape of the curves relating suffix graph measures to string length changes with ω: it is therefore natural to investigate the dependence of these measures on ω. Displaying the relative number of special, antispecial, normal, and terminal points on the same graph reveals a recurrent motif: many strings pass through five phases, marked by the following events (Fig. 21): (1) the increase of special points above terminal points; (2) the increase of normal points above terminal points; (3) the overtaking of special points by normal points; (4) the final overtaking of antispecial points by normal points, after which normal points become the most abundant type in the suffix graph.

Dependence of the relative number of special, antispecial, normal and terminal points on ω. (
A view of the whole datasets (Fig. 22) shows that the values of ω at which each of these transitions occurs is not constant in D1: while there is little variation for the value at which normal points overtake terminal and special points (always around 4,5), and at which special points overtake terminal points (always around 3,4) the value at which normal points overtake antispecial points varies significantly, and it does not respect SCOP boundaries. Superfamily 1.1.1 and fold 2.2 transition at ω ≥ 6, while fold 1.8 either presents no phase transition or transitions at ω = 8. In family 1.8.1.1 normal points never overcome antispecial points, and special and normal points increase above terminal points at relatively high ω. Other exceptions to the pattern occur in D2 where, not surprisingly, DisProt 34 is a good example of abnormality (Fig. 21).
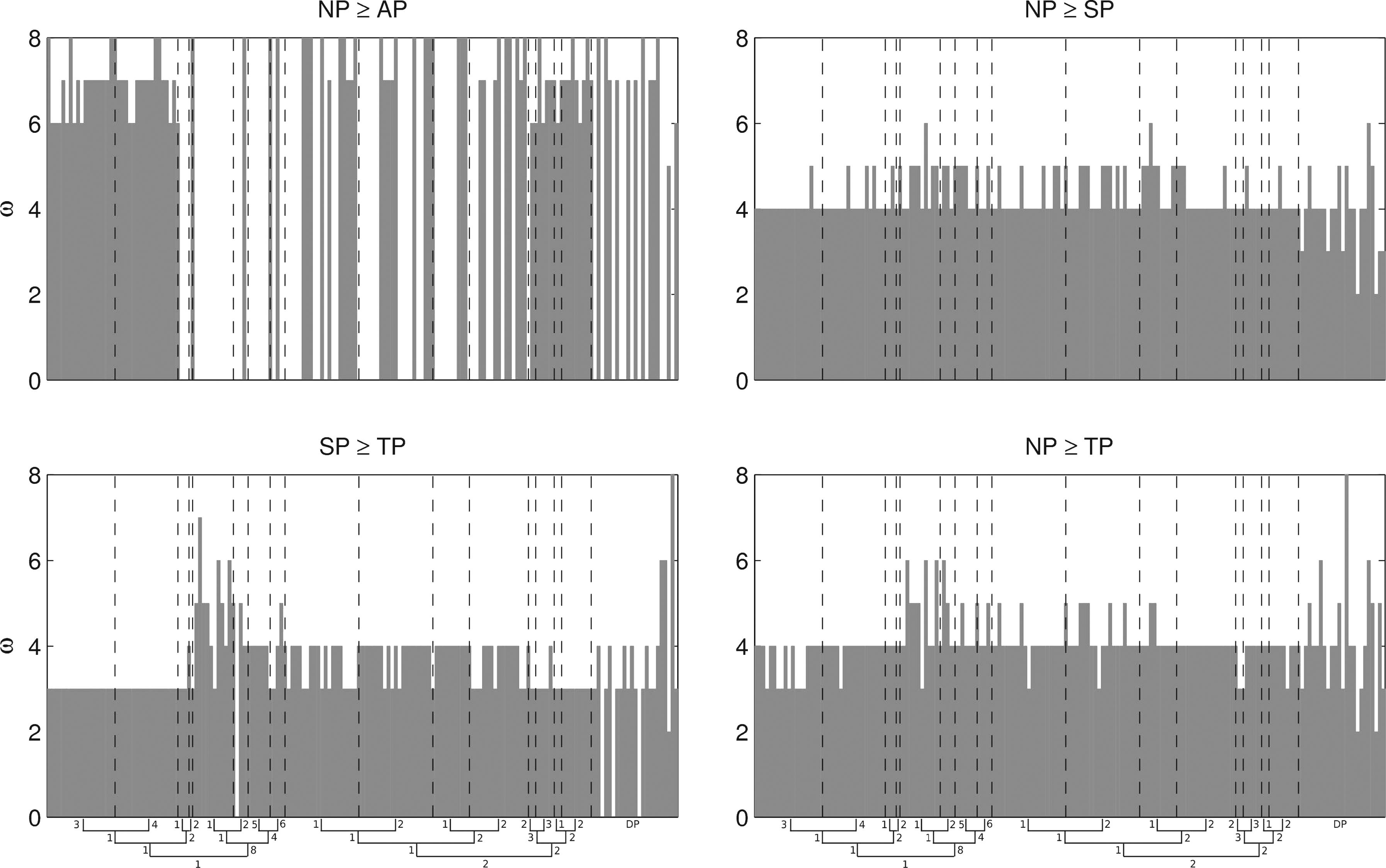
Values of ω at which phase transitions occur in D1 ∪ D2.
This extended heterogeneity prompts us to test whether the relationship between each suffix graph measure and ω is controlled by general rules, like those seen for string length (Fig. 23). It turns out that the relative number of special, antispecial, normal and terminal points trace wide sigmoid bundles in which all strings have similar shape but possibly very different values. 7 These loci are not universal: apart from few obvious outliers in D2 (DisProt 34 in special and normal points, DisProt 13 in special points, DisProt 9 in antispecial and terminal points, DisProt 20 in normal and terminal points), strings in D3 turn out to follow very different rules (Fig. 23). Similar conclusions can be drawn for the absolute number of points and subsequences.

Relative number of points versus ω. (
7. Laws Governing Random Permutations of Polypeptides
The alignment of most polypeptides along the same loci, and the presence of outliers to such loci, proves that the strings in D1 ∪ D2 are not random, but follow a specific compositional pattern, and that this pattern is shared by proteins lying in very different groups of the SCOP hierarchy. We expect the information that encodes these loci to be carried by the sequences of amino acids: in this case, randomly permuting the strings should destroy the signal and cause the loci to degenerate into random clouds. We analyze a set of 100 random permutations for each string in D1 ∪ D2. Surprisingly, it turns out that such permutations still trace the same loci in all graphs of the previous sections (Fig. 24): this proves that, in the dataset analyzed, it is the composition of symbols, not the sequence, to be responsible for the alignment of polypeptides along loci. However, the relationship between distribution of symbols and locus is not bijective: among the members of D1, there are significant variations in how the frequencies of symbols are distributed (Fig. 25); therefore, a locus does not map onto similar distributions of symbols.
Relative number of special points versus string length in D1 ∪ D2 (light gray) and in a copy of D1 ∪ D2 in which each string has been randomly permuted (black).
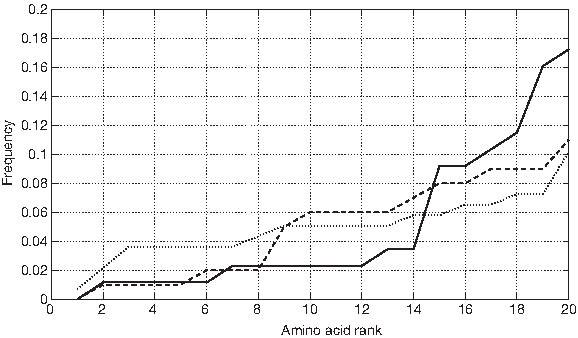
Sorted frequencies of symbols in some strings of D1. (
On the other hand, sequence does influence suffix graph measures: in Figures 12 and 13, for example, random arrangements of a set of 20 symbols with minimum entropy assume a spectrum of different values. We conclude that, in the strings of D1 ∪ D2, the effect of sequence on suffix graph measures is weaker than the effect of symbol composition.
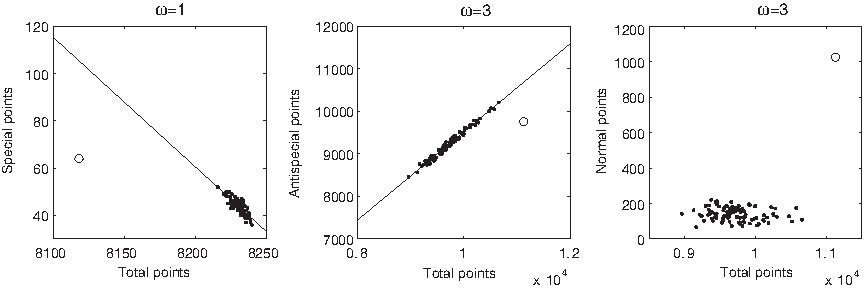
The influence of sequence on suffix graph measures deserves more attention. We project the 100 random permutations of each string in D1 ∪ D2 onto the space generated by every possible (x, y) pair, where x and y are suffix graph measures. Let's concentrate on the relationship between special points and total points first: the visual inspection of the graphs of few strings in D1 shows that the set of random permutations forms a well-defined, linear bundle at ω = 1 and ω ≥ 6, while random clouds appear at 2 ≤ ω ≤ 5 (Fig. 26). Polypeptides are always seen to belong to these bundles.

Number of special points versus number of total points in Hemoglobin I from Scapharca inaequivalvis (a protein in D1, circle) and in 100 random permutations of its sequence (dots). Lines indicate the best linear interpolation of the set of permutations. Other strings in D1 produce similar shapes.
Probing the extent to which this relationship is supported by D1, D2, and D3 is clearly unfeasible by drawing and analyzing each graph visually. Therefore, we measure the correlation coefficient between special and total points for each string: the correlation coefficient represents the strength of linear relationship between two variables as a value between −1 (strong linear negative relationship) and 1 (strong linear positive relationship). We conservatively consider “strong” a correlation that has absolute value ≥0.5 and p-value ≤10−5. The graph of the correlation coefficients for all three datasets (Fig. 27) shows that at ω = 1 the random permutations of all strings have a strong linear negative correlation, except for DisProt 34 and few members of D1 and D3. At ω = 2, 3, 4 the correlation progressively becomes weaker, except in DisProt 34 in which it is strong and positive for all ω > 1 (Fig. 28). At ω ≥ 5 correlation progressively becomes strong and positive for the majority of D1 ∪ D2, but it remains weak in most of D3 (except, for example, thyroid receptor interacting protein 6, Homo sapiens), in some members of D2 (e.g., DisProt 20; Fig. 31), and in fold 1.8 (e.g., in the C-terminal domain of γ, δ resolvase, Escherichia coli). In all cases in which most strings have a strong correlation, strings with a weak correation can be found, and vice versa, proving that the pattern of strong and weak correlations as a function of ω is not an unavoidable regularity of all polypeptides.

Correlation between the number of special and total points in the dataset. For clarity of presentation, p-values are not shown: they are ≤10−5 wherever the correlation is ≥0.5 in absolute value. Vertical dashed lines highlight fold 1.8 and D2. γδR: C-terminal domain of γ, δ resolvase, Escherichia coli. TRIP6: thyroid receptor interacting protein 6, Homo sapiens.

Number of special points versus number of total points in DisProt 34.
The pattern of correlations neither reveals clear distinctions among SCOP groups, nor between polypeptides and their permutations: all strings in the datasets belong to the linear loci of their random permutations, with the exception of DisProt 28, 25 and 34 at ω = 1 (Figs. 28–30). The coefficient a of the linear interpolations is approximately constant inside D1 ∪ D2 ∪ D3 at ω = 1, and approximately constant in D1 ∪ D2 at ω ≥ 6 (Fig. 32). The coefficient b, on the other hand, oscillates widely inside D1 ∪ D2 (Fig. 33).
Number of special (left), antispecial (center), and normal (right) points versus number of total points in DisProt 25.
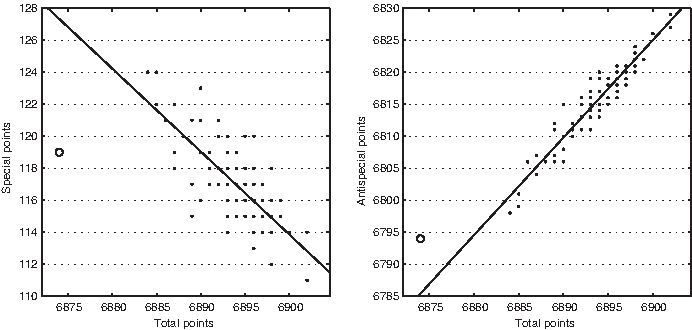
Number of special points (left) and antispecial points (right) versus number of total points in DisProt 28 (ω = 1).
Number of special points versus number of total points in DisProt 20.

Coefficient a of the linear interpolation across the dataset. The coefficient is computed on the measures divided by their respective maximum values. Vertical dashed lines highlight fold 1.8 and D2.
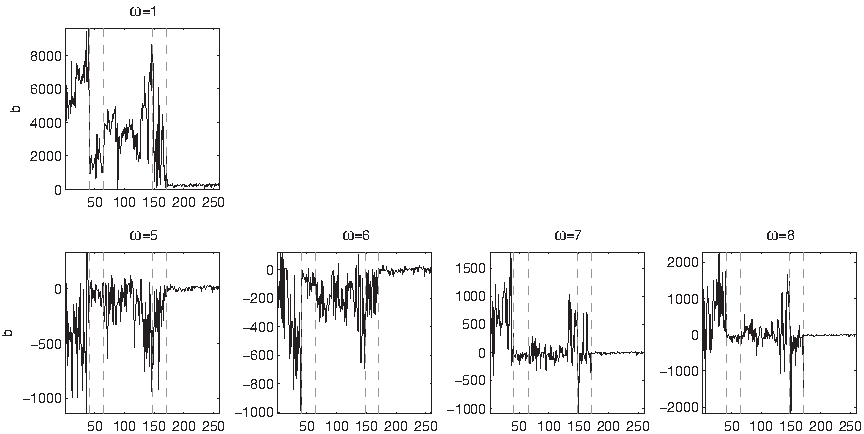
Coefficient b of the linear interpolation across the dataset. The coefficient is computed on the measures divided by their respective maximum values. Vertical dashed lines highlight fold 1.8 and D2.
Analyzing in detail the effect of sequence on each suffix graph measure is outside the scope of this article. Here we just observe that, except for few cases, normal points are not strongly correlated to total points at ω ≤ 4, but at ω ≥ 5 D1 ∪ D2 reaches a strong correlation, while the correlation of D3 remains lower (Fig. 34). Antispecial points are highly correlated with total points at all values of ω; terminal points are not correlated to total points at ω = 1, but they become strongly correlated at ω = 2, 3, and finally their correlation stabilizes around a lower value at ω ≥ 5.
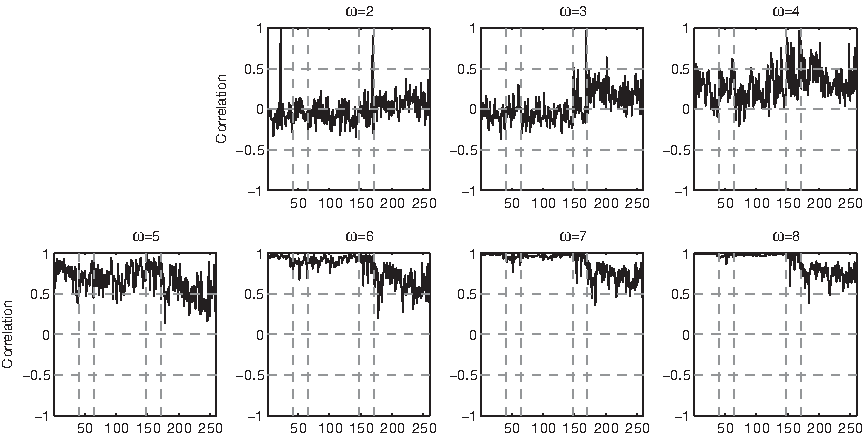
Correlation between the number of normal and total points in the datasets. Vertical dashed lines highlight fold 1.8 and D2.
8. Conclusion
Some natural measures on the abundance of points, arcs, and subsequences in the suffix graphs of polypeptides appear to grasp structural/functional information. The values of these measures depend on string length and on the hiatus of subsequences under a specific set of rules, as shared by a spectrum of structurally and functionally diverse polypeptides. In accordance with the current consensus that sees proteins as random strings, these rules are influenced by the distributions of symbols more strongly than by their organization within the sequences. In most polypeptides, it is seen that even their random permutations amass along specific linear loci. Counterexamples show that none of the rules is an unavoidable property of all polypeptides or distributions of symbols, thereby suggesting that the shapes of these loci are specific signatures of the dataset or of its parts.
It is well known that amino acid composition varies with functional class and cellular localization (Karlin, 1992). The fact that most of the rules described in this paper are common to structurally and evolutionarily diverse polypeptides might suggest that they capture some organizational constraint that crosses the boundaries of protein families, and is implied in the chemical or spatial stability of amino acid chains (Han and Baker, 1995; Han et al., 1997), or in the mechanism by which secondary structures aggregate and connect to each other (Przytycka et al., 2002). Alternatively, these rules might capture evolutionary regularities, like properties of the limited number of peptides that have arguably constituted the primitive peptide world (White and Jacobs, 1990; Qi et al., 2004), or laws behind the assemblage of these original segments into modern domains (Lupas, 2001).
It is also well known that amino acid abundance is highly influenced by the structure of the genome and varies with species (Karlin, 1992). The regularities described in this paper could therefore reflect biases and optimizations in the translation machinery (Schachtel et al., 1991; White, 1994; Dufton; 1997 Wan and Wootton, 2000; Rost, 2002), or be the image of corresponding constraints in the genome.
From the experimental point of view, this work stimulates the analysis of the whole SCOP with the purpose of counting and mapping the repertoire of rules that occur therein. Studying what happens at higher values of ω would also be a natural extension. From the theoretical viewpoint, this work opens the problem of explaining the effect of the sequence and of the distribution of symbols of a string on the described rules. A related avenue consists in defining a complexity measure for strings and distributions hinged on these rules, and in comparing it to other state-vector complexity measures, like Shannon's entropy and the global compositional measure of Wan and Wootton (2000). The problems of computing such measures efficiently and of structurally comparing the suffix graphs of different strings lend themeselves to the formulation of interesting algorithmic extensions.
Footnotes
Acknowledgments
A.A. was supported in part by the Italian Ministry of University and Research (under the Bi-National Project FIRB RBIN04BYZ7), by the United States–Israel Binational Science Foundation (BSF) (grant 008217), and by the Research Program of Georgia Tech. F.C. was supported in part by the Research Program of Georgia Tech and by the A. Gini Foundation, Padova, Italy.
Disclosure Statement
No competing financial interests exist.
1
An exception to this universal rule of disorder is represented by strongly nonrandom polypeptides: about 25% of all amino acids in current databases are estimated to be in “low complexity,” highly regular regions, and about 34% of all proteins in current databases are estimated to contain at least one such low complexity region (Wootton, 1994). These segments are routinely searched for and masked before local alignment searches (Wise, 2001; Jiménez-Montaño, ![]() ).
).
2
For each domain, we use at most three homologues coming from different species. Our choice of branches at each level, and of domains in each class, is arbitrary. For reasons of practical efficiency, domains in D1 have length of 40–200.
3
By “empirical entropy,” we mean the approximation of the entropy of the ergodic source that has generated each sequence using the observed frequency of amino acids.
4
Only the number of normal subsequences is approximately constant at ω = 2, but across the whole D1.
5
When we write that a subtree corresponds to a SCOP group, we mean that the Jaccard coefficient between the subtree and the corresponding SCOP group is at least 0.3, and that there is no other subtree that achieves a significantly larger Jaccard coefficient when compared to the same SCOP group.
6
All the coefficients reported here are computed using the fit function of the Matlab curve fitting toolbox. A detailed investigation of the coefficients of such best interpolations is outside the scope of this paper.
7
There is a tendency for strings in folds 1.1-2.2 to assume a smaller proportion of antispecial and terminal points, and a larger proportion of special and normal points, compared to the rest of D1. We choose not to investigate this detail in the present article.