
Abstract
Abstract
Clusters of genes that have evolved by repeated segmental duplication present difficult challenges throughout genomic analysis, from sequence assembly to functional analysis. These clusters are one of the major sources of evolutionary innovation, and they are linked to multiple diseases, including HIV and a variety of cancers. Understanding their evolutionary histories is a key to the application of comparative genomics methods in these regions of the genome. We propose a probabilistic model of gene cluster evolution on a phylogeny, and an MCMC algorithm for reconstruction of duplication histories from genomic sequences in multiple species. Several projects are underway to obtain high quality BAC-based assemblies of duplicated clusters in multiple species, and we anticipate use of our methods in their analysis.
1. Introduction
In this article, we describe a probabilistic model of evolution of gene clusters on a phylogeny, and devise an algorithm for inference of highly probable duplication histories and ancestral sequences. We apply our algorithm to simulated sequences on the human-chimp-macaque phylogeny, as well as to real clusters assembled from available BAC sequencing data.
Perhaps the first duplication model is due to Fitch (1977), who studied the duplication history of human hemoglobins and apolipoproteins. Inspired by this work, several algorithms were developed for reconstructing histories of gene clusters containing several tandemly repeated copies of a single gene (Benson and Dong, 1999; Tang et al., 2002; Elemento et al., 2002; Zhang et al., 2003). In this model, only tandem duplications are allowed, although recently, Lajoie et al. (2007) investigated model with inversions of variable lengths. However, more complex models are necessary to address evolution of gene clusters in the human genome.
In recent work, genes have been replaced by generic atomic segments (Zhang et al., 2008; Ma et al., 2008; Bertrand et al., 2008). Briefly, a self-alignment is constructed by a local alignment program (e.g., blastz; Schwartz et al., 2003), and only alignments above certain threshold (e.g., 93% for human-macaque split) are kept. Alignment boundaries mark breakpoints, and the sequences between neighboring breakpoints are considered atomic segments (Fig. 1). Under reasonable evolutionary models, sequence similarities between atomic segments are transitive, and the set of atomic segments can be decomposed into equivalence classes, or atom types, such that all segments of the same type have similar sequence. In this way, the nucleotide sequence is transformed into a simpler sequence of atoms.
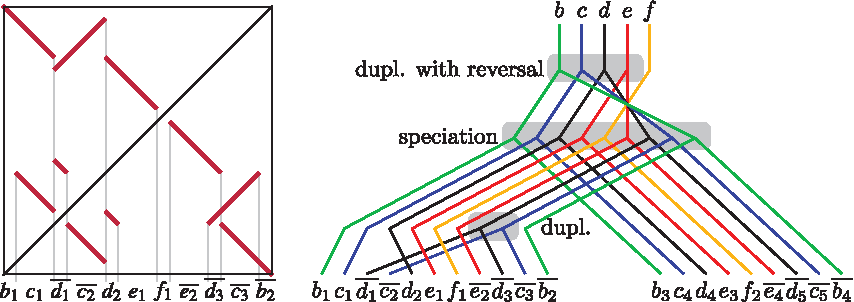
Sequence atomization and segment trees.
The task of duplication history reconstruction is to find a sequence of events (duplications, deletions, and speciations) that starts with an ancestral sequence of atoms, where each atom occurs once, and ends with atomic sequences of extant species. Such a history directly implies segment trees of individual atomic types, implicitly reconciled with the species tree (Fig. 1). Each segment tree represents duplication and speciation events concerning one atom type, similarly as gene tree represents history of a single gene. A common way of looking at these histories is from the most recent events back in time. In this context, we can start from the extant sequences, and unwind events one-by-one, until the ancestral sequence is reached.
Zhang et al. (2008) sought parsimonious solutions of this problems given the sequence from a single species. In particular, they proved a necessary condition to identify candidates for the latest duplication operation, assuming no reuse of breakpoints. After unwinding the latest duplication, the same step is repeated to identify the second latest duplication. Zhang et al. (2008) showed that any sequence of candidate duplications leads to a history with the same number of duplication events under no-breakpoint-reuse assumption. As a result, there may be an exponential number of most parsimonious solutions to the problem, and it may be impossible to reconstruct a unique history. Recently, Zhang et al. (2009) extended the same approach to simultaneous inference in multiple species. A similar parsimony problem has also been recently explored by Ma et al. (2008) in the context of much larger sequences (whole genomes) and a broader set of operations (including inversions, translocations, etc.). In their algorithm, Ma et al. reconstruct a phylogenetic tree for every atomic segment, and reconcile these segment trees with the species tree to infer deletions and rooting. The authors give a polynomial-time algorithm for the history reconstruction, assuming no-breakpoint-reuse and correct atomic segment trees. Both methods make use of extensive heuristics to overcome violations of their assumptions in real data.
The no-breakpoint-reuse assumption is often justified by the argument that in long sequences, it is unlikely that the same breakpoint is used twice (Nadeau and Taylor, 1984). However, there is evidence that breakpoints do not occur uniformly throughout the sequence, and that breakpoint reuse is in fact frequent (Peng et al., 2006; Becker and Lenhard, 2007). Moreover, breakpoints located close to each other lead to short atoms that cannot be reliably identified by sequence similarity algorithms and categorized into atom types. For example, in our simulated data (Section 5), approximately 2% of atoms are shorter than 20bp. These short atoms may appear as additional breakpoint reuses. Thus, no-breakpoint-reuse can be a useful guide, but cannot be relied on in application to real data sets. We have also examined the assumption of correctness of segment trees inferred from sequences of individual segments. For segments shorter than 500 bp (39% of all segments in our simulations) 69% of the trees were incorrectly reconstructed, and even for segments 500–1,000 bp long, a substantial fraction (46%) is incorrect (Fig. 2).
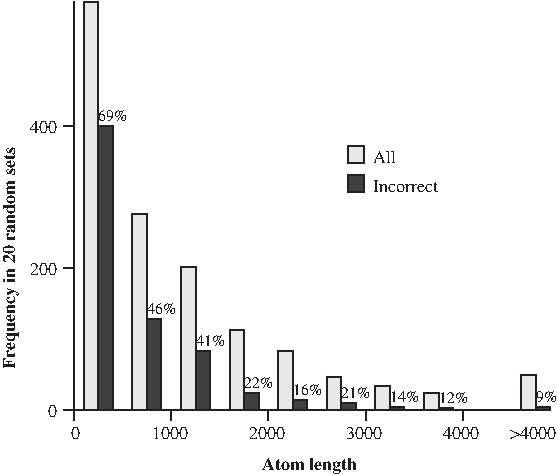
Distribution of atomic segment lengths and accuracy of segment tree inference in 20 simulated fast-evolving clusters (see Section 5). The gray bars show the numbers of segment types. The black bars show the percentages of segment types for which the highest posterior probability unrooted segment tree inferred by MrBayes (Ronquist and Huelsenbeck, 2003) does not match the correct segment tree.
Here, we propose a probabilistic model for sequence evolution by duplication, and we design a sampling algorithm that explicitly accounts for uncertainty in the estimation of segment trees and allows for breakpoint reuse. The results of Zhang et al. (2008) suggest that, in spite of an improved model, there may still be many solutions of similar likelihood. The stochastic sampling approach allows us to examine multiple solutions in the same framework and extract expectations for quantities of interest (e.g., the expected number of events on individual branches of the phylogeny, or local properties of the ancestral sequences).
Our problem is also closely related to the problem of reconstruction of gene trees and their reconciliation with species trees. Even though the recent algorithms for gene tree reconstruction (Wapinski et al., 2007) consider genomic context of individual genes as an additional piece of information, our new methods aim to fully explain genomic context of individual genes through reconstructed duplication histories.
2. Probabilistic Model
In this section, we give a probabilistic model of evolution of gene clusters through segmental duplication on a given species tree T. Later, we use this model for inference of duplication histories and to generate simulated gene clusters.
We start with an ancestral sequence of length N. The sequence evolves by duplications, deletions and substitutions. A duplication copies a source region and inserts the new copy at a target position in the sequence, either on the forward strand (with probability 1 − Pi) or on the reverse strand (with probability Pi). A duplication can be characterized by four coordinates: a centroid (the midpoint of the region between the leftmost and rightmost end of the duplication), the length of the source region, the distance between the source and the target, and a direction (from left to right or from right to left). The centroid is chosen uniformly, and the length and distance are chosen from given distributions (see below). Note that some centroid, distance, and length combinations are invalid; those combinations are rejected. Similarly, a deletion removes a portion of the sequence, and can be characterized by its centroid and length. Each event is a deletion with probability Px, and a duplication with probability (1 − Px). This process straightforwardly defines the probability P(E|len) of any duplication or deletion event E. Here, len is the length of the sequence just before the event E. The number of events on each branch is governed by a Poisson process with rate λ, and thus the probability of observing k events on a branch of length ℓ is Pn(k, ℓ) = (λℓ)ke−λℓ/k!.
A duplication history H generated in this way implies a set σ(H) of atomic segments of several types and a segment tree Tx for each atom type x. The substitutions in the nucleotide sequences of atom x are governed by the HKY substitution model along the segment tree Tx.
We can compute the joint probability p(H, X) of a given set of extant sequences X and a history H (up to a normalization constant) as follows. Let T be a species tree with branches
where P(H, bi) is the probability of events of history H that occur on branch bi of the species tree, Xx represents nucleotide sequences of atoms of type x, and P(Xx|Tx) is the probability of these sequences given tree Txi. For a sequence of events
where len(j − 1) is the length of the sequence before event Ej.
To reduce the number of model parameters, we use geometric distributions to model lengths and distances of duplication events. To estimate these distributions, we have used the lengths and distances estimated by Zhang et al. (2008) from human genome gene clusters (mean length 14,307, mean distance 306,718). The geometric distributions seem to approximate the observed length distributions reasonably well. Similarly, we estimated the probability of duplication with inversion as Pi = 0.39 from the same data, we set the probability of deletion as Px = 0.05, and the length distribution of deletions matches the distribution of duplication lengths. In our model, we do not allow inversions that are not accompanied by a duplication.
Note that for our application, the normalization constant for p(H, X) does not need to be computed. We assume a uniform prior on length distribution of ancestral lengths. This has only a small effect for fixed extant sequences, since the ancestral sequence should contain a single occurrence of each segment type, and therefore the ancestral length is determined mostly by the length of individual atomic segment types. Some combinations of centroids, distances, and lengths will be rejected, but we assume that in long enough sequences, the effect of this rejection step will be negligible and we ignore it altogether.
In the inference algorithm below, we compute likelihood P(Xx|Tx) and branch lengths for each segment tree separately. This independence assumption simplifies computation and allows variation of rates and branch lengths between atoms. This is desirable, since sequences of different functions may evolve at different substitution rates, and selection pressures may change the proportions of individual branch lengths. Nonetheless, branch lengths tend to be correlated among segment trees when individual atoms are duplicated together, and this information is lost by separating the likelihood computations. We are working on a more systematic solution to the problem of rate and branch length variation.
3. Metropolis–Hastings Sampling
For inference of duplication histories, we use the Metropolis–Hastings Markov chain Monte Carlo algorithm (Hastings, 1970) to sample from the posterior probability distribution p(H|X) defined in the previous section, conditional on the extant sequences X and their atomization. The result of the algorithm is a series of samples that can be used to estimate expectations of quantities of interest (e.g., the number of events on individual branches, posteriors of individual segment trees, and particular ancestral sequences), or to examine high likelihood histories.
Briefly, the Metropolis–Hastings algorithm defines a Markov chain whose stationary distribution is the target distribution, but the moves of the Markov chain are defined through a different proposal distribution due to the difficulties of sampling from the target distribution directly. We start by initializing sample history H0. In each iteration, we use a randomized proposal algorithm described below to propose a candidate history
where q(H′|H) is the probability of proposing history H′ if the previous history was H.
The proposal algorithm starts by sampling an unrooted guide tree Tx for every atom type x. The segment trees implied by the proposed history will be later rooted and refined from these guide trees. Guide tree Tx is sampled from the posterior distribution of the trees conditional on a fixed multiple alignment of all instances of atom type x. Sampling guide trees accounts for uncertainty in the estimation of segment trees. We collapse branches with fewer than 5 expected substitutions over the length of the atom sequence, since such short branches usually cannot be estimated reliably. Thus, the guide trees for shorter atoms, where uncertainty is high, will be close to uninformative star trees, while the trees for longer atoms will remain more resolved.
The proposal algorithm samples a history consistent with the given set of guide trees, starting at the leaves of the trees, and progressively sampling groups of atom pairs to merge, until only a single copy of each atom remains. Merging of two groups of atoms corresponds to unwinding one duplication. To obtain a valid history consistent with the guide trees, each of the two groups has to be a contiguous subsequence in the current atomic sequence. Also, the corresponding atoms of the two groups must be of the same type. Finally, the corresponding atoms must be cherries in their guide trees (see also Fig. 3; the leaves xi and xj are cherries in Tx if they have the same parent).
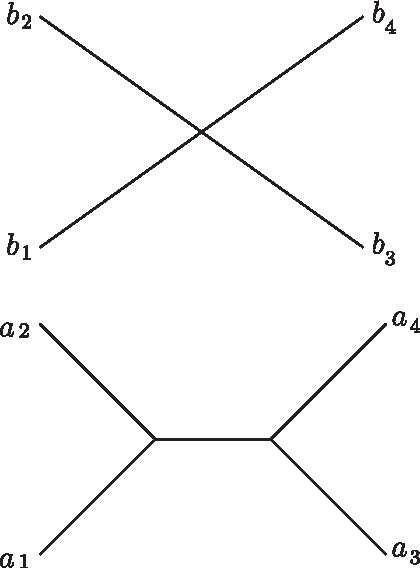
Consistency of histories with the guide trees. Example of guide trees for a sequence of atomic segments a1b1a2b2a3b3a4b4. The segment tree Tb was collapsed into an uniformative star tree, perhaps since atom b is too short. Duplication (a1b1, a2b2) is consistent with the guide trees since (a1, a2) and (b1, b2) are cherries in the corresponding guide trees, but duplication (a1b1, a3b3) is inconsistent, as is (a1b1a2b2, a3b3a4b4).
For example, if the most recent duplication copied atoms a1b1 to atoms a2b2 then a1 and a2 must be cherries in the tree Ta, and b1 and b2 must be cherries in the tree Tb. Unwinding of this duplication will correspond to removal of a2 and b2 from the trees Ta and Tb and from the atomic sequence. Now, the same conditions can be applied to the second latest duplication. In this way, a particular set of guide trees can significantly restrict the set of possible histories.
The sampling distribution over candidate groups of atoms is determined by a series of heuristic penalties described in Section 4, favoring longer duplications, those not inducing breakpoint reuse, and those that were used in the previous sample Hi−1. Even though this algorithm employs a number of heuristics to improve the overall performance of the sampler, they only affect the mixing rate and convergence properties of the sampling, not the asymptotic correctness of the MCMC algorithm.
The multiple alignment for each segment type is created by MUSCLE (Edgar, 2004). Even though it is possible to sample multiple alignments to prevent potential alignment errors from propagating throughout the whole analysis (Holmes and Bruno, 2001), such sampling is by itself computationally intensive. Given that in this paper we consider sequences of greater than 90% similarity, we do not expect multiple alignments to be a major source of error in our reconstructions. Trees, branch lengths, and HKY nucleotide substitution model parameters are sampled by MrBayes (Ronquist and Huelsenbeck, 2003) with uniform prior over tree topologies, and default priors for the other parameters. For each segment type x, all the tree samples are precomputed in a run of 10,000 iterations with a burn-in of 2,500 samples, keeping every 10th sampled tree. In every iteration of the history proposal algorithm, we keep the previous guide tree with 95% probability, otherwise we choose a new tree randomly from the pre-computed samples.
Deletion operations cannot be easily dated, and some of them cannot be even observed in the extant sequence. To address this problem, we attach each deletion to the most recent overlapping duplication or speciation and in the proposal algorithm, described in Section 4, we always propose duplications and corresponding deletions together. To keep the running time feasible, we assume that there is at most one deletion following each duplication and that it does not extend beyond the boundaries of the corresponding duplication segment.
4. Proposal Distribution
As described above, the proposal distribution for histories is defined by a sequential sampling procedure that selects groups of atom pairs to merge in each step. The goal is to define this distribution so that the overall proposal distribution is as close as possible to the actual conditional distribution p(H|X), making the acceptance probability as close as possible to one. Directly characterizing p(H|X) appears to be difficult, so we settle for a heuristic weighting function in the proposal distribution for merges that is designed to produce reasonably good proposed histories. The Metropolis-Hastings algorithm will ensure that the retained samples will accurately reflect the posterior distribution, once the Markov chain reaches stationarity.
In each step, we consider all possible duplications consistent with the current set of guide trees, as well as selected deletion and speciation events. Deletions do not leave observable sequence traces in extant species, and thus it is impossible to date them precisely; instead, in the proposal algorithm we associate deletions with the speciation or duplication events that occurred before them. We allow a single deletion following a duplication. We consider only deletions completely inside the source or target sequence of the duplication.
A speciation event is represented as a copy of all atomic segments from one species to a previously empty sequence of another species, possibly followed by several deletions in both species. We only allow speciations in the partial order imposed by the species tree. Additionally, we propose only speciations that maximize the total sequence length of matched atomic segments between the two species. As in the case of duplications, only segments that are currently cherries in the corresponding guide trees can be matched. For example, if we have sequences in two species S1 = a1b1c1 and S2 = a2b2c2, and b1 and b2 are not cherries in the segment tree, we have to propose speciation from an ancestral sequence ab1b2c or ab2b1c, followed by one deletion in species S1 and one deletion in species S2. Proposals that obey these constraints can be easily generated by a simple dynamic programming algorithm, and in the case of many possible speciation proposals, we only keep 20 highest weight candidates. Note that it is always possible to propose at least one event until we reach an ancestral sequence of unique atoms.
We characterize each proposed event by a feature vector
Target length. The basis of the overall score is the length ℓ of duplication or speciation, i.e. how much sequence is removed by unwinding the event. We set f1 = ln(ℓ) and w1 = 1.
Previously seen event. To keep the newly proposed history similar to the previous sample Hi−1, we add bonus to events seen in Hi−1. This is achieved by a binary indicator feature f2 and weight w2 = ln(10). Some events may not be possible in the new history due to changes in the guide trees.
Branch length mean and variance. For a given duplication consistent with the guide tree set, we can compute the mean distance μ of corresponding cherries in the guide tree (weighted by the lengths of atoms in nucleotides), and also variance on such distance σ. The lower μ indicates likely more recent events, while large variance σ would indicate that we are merging two or more events that happened at different times. We set f3 = μ, f4 = σ, w3 = −10, w4 = −1.
Partial duplication penalty. If the proposed duplication is a subset of a larger duplication, we set indicator f5 = 1 and use w5 = −ln(100).
Breakpoint reuse penalty. Although, we allow breakpoint reuse, we favor duplications with fewer breakpoint reuses which seems to be particularly useful for determining correct direction of duplications. We have implemented the three conditions stipulated by Zhang et al. (2008) based on collapsibility of atom pairs on boundaries of the duplicated segments. We set f6 to the number of violated conditions and w6 = −ln(10);
Pair reduction bonus. Consider the number π of distinct pairs of adjacent atom types that occur in the current set of sequences. For input with n atom types, π = n − 1 when we reach the ancestral sequence, and each duplication reduces π by at most 2. This gives us a lower bound on the number of events necessary to reach the ancestral sequence. We set f7 to be the reduction of π achieved by the event ( f7 can be negative if π increases) and w7 = ln(10).
Deletion penalties. Deletion associated with a duplication is penalized by setting f8 = 1 and w8 = −ln(10). In addition, we penalize longer deletions by setting f9 = ln(d/(d + ℓ)) and w9 = 3 where ℓ is the length of the target sequence in the duplication, and d is the length of the deletion. Each deletion associated with a speciation is penalized by setting f10 = 1 and w10 = −ln(1000).
Heat constants. Finally, in some rounds of the MCMC sampler, we want to explore radically new histories, while in other rounds we want to concentrate on smaller local improvements. Thus, we exponentiate the final event weights to a heat constant, which changes from round to round. In our experiments, we have used cyclic sequence of heats (0.5, 0.6, 1, 1.2).
5. Experiments
We have implemented the MCMC sampler described above and verified its functionality on simulated data. For the simulations, we have estimated branch lengths and HKY model parameters (equilibrium frequencies and transition/transversion ratio) from the UCSC syntenic alignments (Karolchik et al., 2008) of human, chimp, and macaque on human chromosome 22.
We created 20 simulated gene clusters in each of the following two categories: slow evolving and fast evolving (duplication rate at 200 and 300 times substitutions per site, respectively). We have applied our algorithm to atomic segments derived from the simulation, with short (<500bp) atomic segments removed to emulate the increase in breakpoint reuse due to imperfect identification of alignment boundaries in real data sets (Table 1 for the data set overview). For each cluster, we ran two chains of the sampler from random starting points for up to 10,000 iterations each, discarding the first 2,500 samples as burn-in. The sampler seems to converge reasonably quickly (Fig. 4).
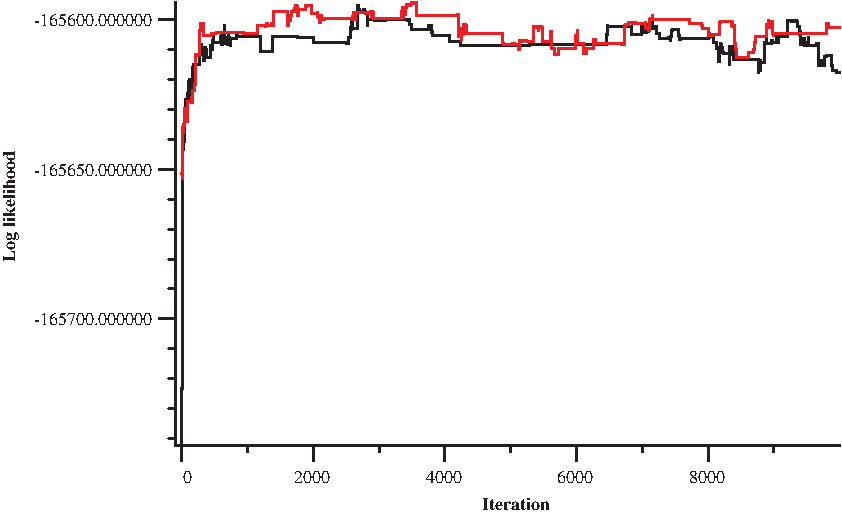
Convergence of the MCMC sampler. Log likelihood as a function of iteration number for two independent chains with random starting points on a slowly evolving simulated cluster.
In the majority of cases, we predict the correct number of events (Table 2; 14 out of 20 for slowly evolving, 15 out of 20 for fast evolving clusters). Note that in some cases the predicted number of events is lower than the actual number of events: this is likely due to events that become invisible in the extant sequences because of subsequent deletions. We have compared our results on the human lineage to Zhang et al. (2008), and on the whole tree to Zhang et al. (2009). The performance has improved, especially in the case of fast evolving clusters. We also examined the correctness of distribution of events along the phylogeny (Table 3). Finally, we compared the predicted and actual ancestral atomic sequences. To quantify the differences between the sequences, we have counted the number of breakpoints required to transform the predicted ancestral sequence to the actual ancestor (Fig. 5). In the majority of cases (31 out of 40), the expected number of breakpoints is smaller than 0.5.

Histogram of expected number of incorrect breakpoints on the 40 simulated data sets. The number of breakpoints required to transform predicted sequences to actual sequences is computed over all MCMC samples and the average is rounded to the closest integer.
The table shows the histogram of differences between the real number of events and the predicted number of events along the human lineage and on the whole tree on the 40 simulated data sets (20 with slow duplication rate 200, 20 with fast duplication rate 300). MCMC: rounded expected number of events from all samples. ML: highest likelihood sample. We compare to results of Zhang et al. (2008) on single species (Z2008) and Zhang et al. (2009) on the whole tree (Z2009). Note that the results of the two programs are not directly comparable, since our program was run on correct atomization with short atoms filtered out (giving Z2008 and Z2009 advantage of smaller amount of breakpoint reuse in the data), while Z2008 and Z2009 used their own built-in atomization method (giving advantage to our program, since the results of their atomization may be potentially incorrect).
The table shows a histogram of the differences between the actual and the expected number of events computed from the MCMC samples.
Beyond the simulated data, we have applied our algorithm to the following gene cluster sequences: PRAME (human-macaque phylogeny), AMY (human-macaque phylogeny), and UGT1A (human-chimp-orang phylogeny). PRAME cluster (preferentially expressed antigen in melanoma) is one of the most active gene clusters in the human genome, and shows strong evidence of positive selection (Birtle et al., 2005; Gibbs et al., 2007). AMY cluster contains five amylase genes that are responsible for digestion of starch. It appears to have expanded much faster in humans than in other primates, according to aCGH experiments (Dumas et al., 2007). The UGT1A cluster consists of multiple isoforms of a single gene, instrumental in transforming small molecules into water-soluble and excretable metabolites. This gene has at least thirteen unique alternate first exons resulting from duplications at various stages of mammalian evolution. UGT1A provides an unusual opportunity for studying promoter evolution.
Recently duplicated clusters are grossly missassembled in shotgun based genomes (Green, 2001; Zhang et al., 2008). To prepare our data sets, we have first screened sequenced BACs from chimp, orangutan, and macaque for similarity with the corresponding human sequences, assembled BACs into longer contigs, and selected subregions whose ends showed clear homology with upstream and downstream sequences of the human cluster. To identify atomic segments, we divide the sequences into equally sized 500 bp windows, and for each window we find approximate copies in all available sequences at 90% identity cutoff. The atoms are assigned in a greedy way (starting from the windows with the largest number of copies), and windows overlapping already assigned atoms are discarded. Finally, atoms that always occur in pairs are merged into longer atoms. Table 1 shows an overview of the resulting sequences and atoms.
For each cluster, we ran five chains from different starting points for 5,000–10,000 samples, discarding the first 2,500 samples. We have estimated the number of duplications and deletions overall (Table 1), and on individual branches of the phylogeny (Fig. 6). The estimated numbers of duplications for PRAME and AMY are comparable to those of Zhang et al. (2008). With UGT1A, we obtain higher estimates possibly due to differences in our atomization procedure or effects of the additional species in the analysis.

Estimated numbers of events. For each cluster, we show the posterior mean and standard deviation of the number of duplications (above the branch) and deletions (below the branch) as assessed by MCMC sampling. The root branch shows events up until 90% sequence similarity cutoff.
Figure 7a shows the highest likelihood reconstruction of the history of the UGT1A cluster. The cluster contains several isoforms of the same five-exon gene. Exons 2–5 are shared among all the isoforms, while exon 1 is alternatively spliced. The reconstruction shows division of the first exons into three distinct groups, and their ortholog/paralog relationships in human, chimp, and orangutan.
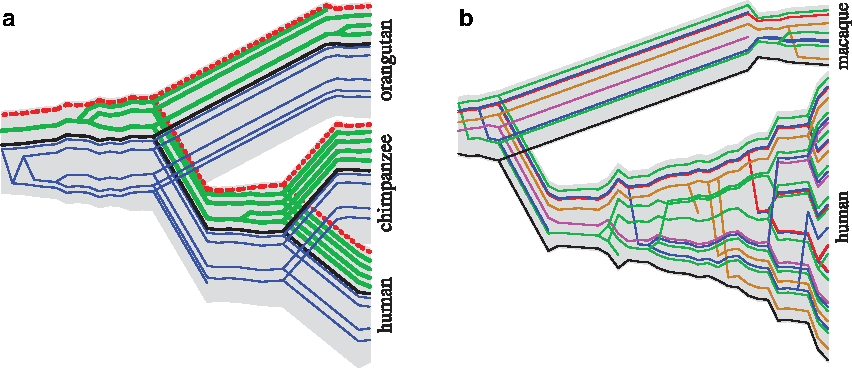
Highest likelihood reconstruction of duplication histories. The branch lengths in the figures do not correspond to the actual branch lengths. The atoms are ordered in their order along the genomic sequences (extant and ancestral).
While the duplication history of the UGT1A cluster consists of mostly ancient events, the PRAME cluster (Fig. 7b) shows recent large-scale duplications, especially in the human lineage. Figure 7 shows such events by several co-linear bifurcations at the same level of the tube tree. The reconstruction of the history by traditional methods (gene tree/species tree reconciliation) is complicated by the presence of recent duplications (99% similarity), and chimeric genes (Gibbs et al., 2007). We address these issues by considering multiple guide trees for each atom as well as spatial configuration of atoms in multiple species. However, the predicted history is by no means perfect. Rhesus sequence exhibits large regions that apparently arose by a single event, yet we split this event due to mistakes in atomic representation. We expect that improved procedure for segmenting sequence into atoms will address this problem.
Figure 8 shows a cartoon of the ancestral atomic PRAME sequence of the highest posterior probability, as well as other pairs of adjacent atoms with posterior probability of >25%. This reconstruction of ancestral atomic sequence shows more uncertainty than similar reconstructions in the simulated data, though there are large blocks resolved uniquely.

Ancestral sequence reconstruction for PRAME. The cartoon shows large blocks of consecutive atomic segments, with block size proportional to the number of atoms per block. The blocks are ordered according to the highest posterior ordering and the alternative edges show other possible pairs of adjacent atoms with >25% posterior probability. The atoms spanning five ancestral genes at 90% similarity are marked A–E.
6. Discussion
In this article, we have introduced a new model of evolution of gene clusters and designed an algorithm to reconstruct high probability evolutionary histories of these clusters. We have tested our method on both simulated and real data. Comparative genomics methods traditionally concentrate on sequences where 1:1 orthology can be established. In case of gene clusters, this is rarely the case. Our efforts in reconstruction of gene cluster histories will support further development of comparative genomic tools to analyze these complex regions.
Gene clusters should not be seen only as a confounding factor. The number of orthologous sequences, their divergence, and phylogenetic relationships greatly impact the accuracy of comparative genomic studies. For example, Kosiol et al. (2008) has shown that the sensitivity of positive selection scans is improved by considering sequences from a complex phylogeny. Studies based on orthologous regions between species can at present use a phylogeny of up to 10 orthologous copies of a particular mammalian gene from genomes sequenced at reasonable quality. On the other hand, some clusters contain many more copies with significantly more complex phylogeny even within a single species (e.g., the PRAME cluster contains more than 30 copies in the human genome alone). Thus, the gene clusters provide an opportunity for refined look at evolution of genes and genomes. Multiple sources of evidence suggest that many interesting developments in genomes happen within the boundaries of gene clusters, which further increases interest in their study. Multiple efforts are currently under way to BAC sequence selected gene clusters in multiple species and in multiple populations (Zhang et al., 2008; Zody et al., 2008). Accurate methods and models for reconstruction of duplication histories of these clusters are essential in understanding the evolution, function, and biomedical implications of these regions.
The general framework of our method allows future developments. One limitation of our sampler is its low sample acceptance ratio, indicating low level of mixing in the Markov chain. We plan to devise a systematic way for tuning the parameters in the proposal distribution towards better acceptance ratios. We also plan to improve the underlying probabilistic model. Currently the branch lengths in segment trees are chosen independently of the duplication history. Instead, we plan to consistently date duplication events on each branch, and use a scaling parameter for each atom type so that we can accurately model correlation between branch lengths of individual atom types and at the same type allow rate variation in different parts of the sequence. We use a simple HKY substitution model with variance in rates allowed between individual atomic segments. In future work, it will be possible to employ more complex models of sequence evolution, such as variable rate site models and models of codon evolution, within the same framework. Such extensions will allow us to identify sites and branches under selection in gene clusters in a principled way, and contribute towards better functional characterization of these important genomic regions. An interesting alternative approach might be to use combinatorial optimization instead of sampling to find maximum likelihood history in the above model.
Footnotes
Acknowledgments
We would like to thank Devin Locke and LaDeana Hillier at Washington University, St. Louis for providing us with the BAC sequences from chimp, orangutan, and macaque. We would also like to thank Webb Miller and Yu Zhang for helpful discussions on this problem. Research of T.V. and B.B. is supported by European Community FP7 grants IRG-224885 and IRG-231025 and by a grant from VEGA (1/0210/10). Research of A.S. is supported by U.S. National Science Foundation grant DBI-0644111.
Disclosure Statement
No competing financial interests exist.