
Abstract
Abstract
A binary matrix has the Consecutive Ones Property (C1P) if its columns can be ordered in such a way that all 1's on each row are consecutive. A Minimal Conflicting Set is a set of rows that does not have the C1P, but every proper subset has the C1P. Such submatrices have been considered in comparative genomics applications, but very little is known about their combinatorial structure and efficient algorithms to compute them. We first describe an algorithm that detects rows that belong to Minimal Conflicting Sets. This algorithm has a polynomial time complexity when the number of 1s in each row of the considered matrix is bounded by a constant. Next, we show that the problem of computing all Minimal Conflicting Sets can be reduced to the joint generation of all minimal true clauses and maximal false clauses for some monotone boolean function. We use these methods on simulated data related to ancestral genome reconstruction to show that computing Minimal Conflicting Set is useful in discriminating between true positive and false positive ancestral syntenies. We also study a dataset of yeast genomes and address the reliability of an ancestral genome proposal of the Saccharomycetaceae yeasts.
1. Introduction
If the matrix M contains only correct information (i.e., groups of markers that were co-localized in the ancestral genome of interest), then it has the C1P, which can be decided in linear-time and space (Booth and Lueker, 1976; Habib et al., 2000; Hsu, 2002; McConnell, 2004; Meidanis et al., 1998). However, with most real datasets, M contains errors. These can be either incorrect columns, that represent genomic markers that were not present in G, or incorrect rows, that represent groups of markers that were not co-localized in G.1 A fundamental question is then to detect such errors in order to correct M, and the classical approach to handle these (unknown) errors relies on combinatorial optimization, asking for an optimal transformation of M into a matrix that has the C1P, for some notion of transformation of a matrix linked to the expected errors; for example, if incorrect markers (resp. groups of co-localized genes) are expected, one could ask for a maximal subset of columns (resp. rows) of M that has the C1P. In both cases, such combinatorial optimization problems are intractable (Dom, 2008, 2009) for recent surveys.
In the present work, we assume the following situation: M is a binary matrix that represents information about an unknown ancestral genome G and does not have the C1P due to erroneous rows, from now called false positives. The notion of Minimal Conflicting Set was introduced to handle non-C1P binary matrices and false positives in Bergeron et al. (2004) and Stoye and Wittler (2009). If a binary matrix M does not have the C1P, a Minimal Conflicting Set (MCS) is a submatrix M′ of M composed of a subset of the rows of M such that M′ does not have the C1P, but every proper subset of rows of M′ has the C1P. The Conflicting Index (CI) of a row of M is the number of MCS it belongs to. Hence, MCS can be seen as the smallest structures that prevent a matrix from having the C1P. It is then natural to expect that false positive belong to MCS, and that every MCS contains at least one false positive. In Bergeron et al. (2004), an extreme approach was followed in handling non-C1P matrices: all rows belonging to at least one MCS were discarded from M, which can consequently discard also a large number of true positives. In Stoye and Wittler (2009), rows were ranked according to their CI (or more precisely an approximation of their CI) before being processed by a branch-and-bound algorithm to extract a maximal subset of rows of M that has the C1P. These two approaches raise natural algorithmic questions related to MCS that we address here:
For a row r of M, is the CI of r greater than 0? Can we compute the CI of all rows r of M or enumerate all MCS of M?
Our work is motivated by the fact that the fundamental question is to detect the false positives rows in M rather than extracting a maximal C1P submatrix. We investigate here, using both simulations and real data, the following question: does a false positive row have some characteristic properties in terms of MCS or CI? This question naturally extends to the notion of Maximal C1P Sets (MC1PS), a dual notion of MCS, that represent sets of row that do have the C1P but can not be extended while maintaining this property.
After some preliminaries on the C1P, MCS, and MC1PS (Section 2), we attack two problems. First, in Section 3, we consider the problem of deciding if a given row of a binary matrix M belongs to at least one MCS. We show that, when all rows of a matrix are constrained to have a bounded number of 1's, deciding if the CI of a row of a matrix is greater than 0 can be done in polynomial time. The constraint on the number of 1's per row is motivated by real applications: in Ma et al. (2006), for example, adjacencies, that is rows with two 1s per row, were considered for the reconstruction of an ancestral mammalian genome. Next, in Section 4, we attack the problem of generating all MCS or MC1PS for a binary matrix M. We show that this problem can be approached as a joint generation problem of minimal true clauses and maximal false clauses for monotone boolean functions. This can be done in quasi-polynomial time thanks to an oracle-based algorithm for the dualization of monotone boolean functions (Eiter et al., 2008; Fredman and Khachiyan, 1996; Gurvich and Khachiyan, 1999). We (U.-U. Haus and T. Stephen) implemented this algorithm and applied it on simulated and real data (Section 5). Application on simulated data suggest that computing all conflicting sets and the conflicting index of all rows of a binary matrix is useful to discriminate between true positive and false positive ancestral syntenies. We also study a real dataset of yeast genomes and address the reliability of an ancestral genome proposal of the Saccharomycetaceae yeasts. We conclude by discussing several open problems.
2. Preliminaries
We briefly review here ancestral genome reconstruction and known algorithmic results related to Minimal Conflicting Sets and Maximal C1P Sets.
2.1. The consecutive ones property, minimal conflicting sets, maximal C1P sets
Let M be a binary matrix with m rows and n columns, with e entries 1. We denote by
Definition 1
A Minimal Conflicting Set (MCS) is a set R of rows of M that does not have the C1P but such that every proper subset of R has the C1P. The Conflicting Index (CI) of a row ri of M is the number of MCS that contain ri. A row ri of M that belongs to at least one conflicting set is said to be a conflicting row. The Conflicting Ratio (CR) of a row ri of M is the ratio between the CI of ri and the number of MCS that M contains.
Definition 2
A Maximal C1P Set (MC1PS) is a set R of rows of M that has the C1P and such that adding any row from M to R results in a set of rows that does not have the C1P. The MC1PS Index (C1PI) of a row ri of M is the number of MC1PS that contain ri. The MC1PS Ratio (C1PR) of a row ri of M is the ratio between the C1PI of ri and the number of MC1PS that M contains.
For a subset
2.2. Ancestral genome reconstruction
The present work is motivated by the problem of inferring an ancestral genome architecture given a set of extant genomes. An approach to this problem, described in Chauve and Tannier (2008), consists in defining an alphabet of genomic markers that are believed to appear uniquely in the extinct ancestral genome. An ancestral synteny is a set of markers that are believed to have been consecutive along a chromosome of the ancestor. A set of ancestral syntenies can then be represented by a binary matrix M: columns represent markers and the 1 entries of a given row define an ancestral synteny. If all ancestral syntenies are true positives (i.e. represent sets of markers that were consecutive in the ancestor), then M has the C1P and defines a set of Contiguous Ancestral Regions (CARs) (Chauve and Tannier, 2008; Ma et al., 2006). Otherwise, some ancestral syntenies are false positives that create MCS and the key problem is to detect and discard them.
Note however that we do not assume that any false positive creates an MCS; indeed it is possible that a false positive row contains two entries 1 corresponding to two markers that are extremities of two real ancestral chromosomes, and then this false positive would create a chimeric ancestral chromosome. Such false positives have to be detected using techniques other than the ones we describe in the present work, as there is no combinatorial signal to detect them based on the Consecutive Ones Property.
In the framework described in Chauve and Tannier (2008) and also used in Adam et al. (2007), Ma et al. (2006), and Stoye and Wittler (2009), ancestral syntenies are defined as common intervals: markers consist of two genome segments having the same content (Bergeron et al., 2008) of a pair of genomes whose evolutionary path goes through the desired ancestor. As ancestral syntenies are detected by mining common intervals in pairs of genomes, it is then easy to control the degree of the resulting matrix by restricting the comparison to common intervals of bounded size, such as adjacencies (ancestral syntenies of size 2), which is fundamental for the result we describe in Section 3.
Also, it is common to weight the rows of M with a measure of confidence in its quality, for example based on the distribution of a group of co-localized markers among the considered extant genomes (Chauve and Tannier, 2008; Ma et al., 2006), and we can expect that false positives have a lower score in general than true positives. However, the concepts of MCS and MC1PS are independent of this weighting and we do not consider it here from a theoretical point of view, although we do consider it in our experiments on real data.
2.3. Preliminary algorithmic results on MCS and MC1PS
In the case where each row of M has exactly two entries 1, M naturally defines a graph GM with vertex set
Property 1
If Δ(M) = 2, a set of rows R of M is an MCS if and only if the subgraph induced by the corresponding edges is a star with four vertices (also called a claw) or a cycle.
This property implies immediately that both the number of MCS and the number of MC1P can be exponential in n. Also, combined with the fact that counting the number of cycles that contain a given edge in an arbitrary graph is #P-hard (Valiant, 1979), this leads to the following result.
Theorem 1
The problem of computing the Conflicting Index of a row in a binary matrix is #P-hard.
Given a set of p rows R of M, deciding whether these rows form an MCS can be achieved in polynomial time by testing (1) whether they form a matrix that does not have the C1P and, (2) whether every maximal proper subset of R (obtained by removing exactly one row) forms a matrix that has the C1P. This requires only p + 1 C1P tests and can then be done in time O(p(n + p + e)), using an efficient algorithm for testing the C1P (McConnell, 2004).
The problem of generating one MCS is not hard and can be achieved in polynomial time by the following simple greedy algorithm:
let for i from 1 to m, if removing ri from R results in a set of rows that has the C1P then keep ri in R, otherwise remove ri from R; the subset of rows R obtained at the end of this loop is then an MCS.
Given M and a list Let M′ be defined by removing from M at least one row from each Ri in C by recursing on the elements of C. If M′ does not have the C1P then compute an MCS of M′ and add it to C, else backtrack to step 1 using another set of rows to remove such that each
This algorithm can require time Ω(nk) to terminate, which, as k can be exponential in n, can be superexponential in n. As far as we know, this is the only previously proposed algorithm to compute all MCS.
Remark 1
The algorithms to decide if a set of rows is an MCS, compute an MCS or compute all MCS can be transformed in a straightforward way to answer the same questions for MC1PS, with similar complexity. The analogue of Property 1 for MC1PS in matrices of degree 2 is that a MC1P is a maximal set of paths: adding an edge creates a claw or a cycle. We are not aware of any result on counting or enumerating maximal sets of paths.
In summary, the number of MCS or MC1PS can be exponential, and there is no known efficient algorithm to decide, in general, if a given row belongs to some MCS. In the next section we show that there is an efficient algorithm if Δ(M) is fixed.
3. DECIDING IF A ROW IS A CONFLICTING ROW
We now describe our first result, an algorithm to decide if a row of M is a conflicting row (i.e., has a CI greater than 0). Detecting non-conflicting rows is important, for example to speed-up algorithms that compute an optimal C1P subset of rows of M, or in generating all MCS. Our algorithm has a complexity that is exponential in Δ(M). It is based on a combinatorial characterization of non-C1P matrices due to Tucker (1972).
Tucker patterns. The class of C1P matrices is closed under column and row deletion. Hence there exists a characterization of matrices which do not have the C1P by forbidden minors. Tucker (1972) characterizes these forbidden submatrices, called MI, MII MIII, MIV, and MV: if M is binary matrix that does not have the C1P, then it contains at least one of these matrices as a submatrix. We call these forbidden matrices the Tucker patterns. Patterns MIV and MV each have 4 rows and respectively 6 and 5 columns, while MI,MII,MIII are (q + 2) by (q + 2), (q + 3) by (q + 3) and (q + 2) by (q + 3), respectively, for a parameter q ≥ 1. When q = 1, pattern MI corresponds to cycle and pattern MIII corresponds to the claw. The patterns are described in the Appendix.
Bounded patterns. Let P be a set of p rows R of M, that defines a p × n binary matrix. P is said to contain exactly a Tucker pattern MX if a subset of its columns defines a matrix equal to pattern MX. The following properties are straightforward from the definitions of Tucker patterns and MCS:
Property 2
(1) A set P of rows of M is an MCS if and only if P contains exactly a Tucker pattern and no proper subset of rows of P does contain exactly a Tucker pattern. (2) If a subset of p rows of M contains exactly a Tucker pattern MII MIII, MIV or MV, then 4 ≤ p ≤ max(4,Δ(M) + 1).
If a row ri satisfies the conditions of Property 2.(1) for a Tucker pattern MX, we say that ri belongs to an MCS due to pattern MX. Tucker patterns with at most Δ(M) + 1 rows are said to be bounded. Property 2 leads to the following results.
Proposition 1
Let M be a binary matrix that does not have the C1P, and ri a row of M. Deciding if ri belongs to an MCS due to a Tucker pattern of p rows can be done in O(mp−1p(n + p + e)) worst-case time.
Proof
The following brute-force algorithm decides if ri belongs to an MCS due to a Tucker pattern of p rows.
Examine all sets of p rows of M that contain ri. For a given set of rows, if it does not have the C1P but every proper subset does have the C1P, then ri belongs to an MCS due to a Tucker pattern of p rows. If no such set satisfies this property, then ri does not belong to any MCS due to a Tucker pattern of p rows.
The complexity can be explained as follows: there are O(mmax(3,p−1)) subsets of the m rows of M to consider. For each such set, we need to perform at most p + 1 C1P tests, and each such test can be performed in O(n + p + e) worst-case time. ▪
The following corollary follows immediately from Property 2.(2).
Corollary 1
Let M be a binary matrix that does not have the C1P, and ri a row of M. Deciding if ri belongs to an MCS due to a bounded Tucker pattern can be done in O(mmax(3,Δ(M))Δ(M)(n + Δ(M) + e)) worst-case time.
Unbounded patterns. We now describe how to decide if a row ri of M belongs to an MCS due to an unbounded Tucker pattern. From Property 2.(2), this Tucker pattern can only be a pattern MI with at least Δ(M) + 2 rows. The key idea is that Tucker pattern MI describes a cycle in a bipartite graph encoded by M.
Let BM be the bipartite graph defined by M as follows: vertices are rows and columns of M, and every entry 1 in M defines an edge. Pattern MI with p rows corresponds to a cycle of length 2p in BM. Hence, if R contains MI with p-row, the subgraph of BM induced by R contains such a cycle and possibly other edges.
Let
Proposition 2
Let M be a binary matrix that does not have the C1P and ri be a row of M that does not belong to any MCS due to a bounded Tucker pattern. Then ri belongs to an MCS if and only if ri belongs to a cycle
Proof
If ri belongs to an MCS but not due to a bounded Tucker pattern, then, according to Property 2.(2), it is due to pattern MI defined on p rows of M containing ri. So ri belongs to a cycle C defined by this pattern MI. However, if ri is blocked in C, then, removing the row ri and its adjacent edges still leaves a cycle in the subgraph of BM induced by the remaining vertices, which contradicts the fact that the initial p rows form an MCS.
Now, assume that ri belongs to a cycle The set We now want to show that removing any row from this gives a matrix that has the C1P. Let
The algorithm. To decide whether a row ri of M belongs to an MCS, we can then use the following algorithm.
Decide whether it belongs to an MCS due to a bounded pattern as follows: (a) Enumerate all subsets of at least 4 and at most Δ(M) + 1 rows of M that contain ri. (b) For each such subset, if it is not C1P but all its proper subsets are C1P, then ri belongs to an MCS. If ri does not belong to an MCS due to a bounded pattern then (a) Enumerate all pairs of rows (b) For each such pair
This algorithm proves the main result of this section:
Theorem 2
Let M be an m × n binary matrix that does not have the C1P, and ri be a row of M. Deciding if ri belongs to at least one MCS can be done in O(mmax(3,Δ(M))Δ(M)(n + Δ(M) + e)) time.
4. Generating All MCS and MC1PS Using Monotone Boolean Functions
In this section, we describe an algorithm that enumerates all MCS and MC1PS of a binary matrix M simultaneously in quasi-polynomial time. The key point is to describe this generation problem as a joint generation problem for monotone boolean functions.
Let
Definition 3
A boolean function f: {0, 1}
m
→ {0, 1} is said to be monotone if for every
Definition 4
Given a boolean function f, a boolean vector X is said to be a Minimal True Clause (MTC) if f (XI) = 1 and f (XJ) = 0 for every
For a given m × n binary matrix M, let fM: {0, 1}
m
→ {0, 1} be the boolean function defined by fM(XI) = 1 if and only if RI does not have the C1P, where
Proposition 3
Let
It follows from Proposition 3 that generating all MCS reduces to generating all MTC for a monotone boolean function. This very general problem has been the subject of intense research, and we briefly describe below some important properties.
Theorem 3 (Gurvich and Khachiyan 1999)
Let
Theorem 4 (Gunopulos et al., 1997)
The problem of generating all MTC of a monotone boolean function f using an oracle to evaluate this function can require up to |MTC( f ) + MFC( f)| calls to this oracle.
This property suggests that, in general, to generate all MTC, it is necessary to generate all MFC, and vice-versa. For example, the algorithm of Stoye and Wittler (2009) described in Section 2 is a satisfiability oracle based algorithm—it uses a polynomial-time oracle to decide if a given submatrix has the C1P, but it doesn't use this structure any further. Once it has found the complete list C of MCS, it will proceed to check all MC1PS sets as candidate conflicting sets before terminating. Since this does not keep the MC1PS sets explicitly, but instead uses backtracking, it may generate the same candidates repeatedly resulting in a substantial duplication of effort. In fact, this algorithm can easily be modified to produce any monotone boolean function given by a truth oracle.
One of the major results on generating MTC for monotone boolean functions, is due to Fredman and Khachiyan. It states that generating both sets together can be achieved in time quasi-polynomial in the number of MTC plus the number of MFC.
Theorem 5 (Fredman and Khachiyan, 1996)
Let f: {0, 1}
m
→ {0, 1} be a monotone boolean function whose value at any point
The key element to achieve this result is an algorithm that tests if two monotone boolean functions are duals of each other (Eiter et al., 2008). As a consequence, we can then use the algorithm of Fredman and Khachiyan to generate all MCS and MC1PS in quasipolynomial time.
5. Experimental Results
We present here results obtained on simulated and real datasets using the cl-jointgen implementation (release 2008-12-01) of the joint generation method which is publicly available (from U.-U. Haus and T. Stephen) with an oracle to test the C1P property based on the algorithm described in McConnell (2004).
5.1. Simulated data
We generated several simulated datasets of ancestral syntenies as follows:
We started from an ancestral unichromosomal genome G composed of 40 genomic markers, labeled from 1 to 40 and ordered increasingly along this chromosome (i.e. it is represented by the identity permutation on From this ancestor, we extracted 39 true positive ancestral syntenies, labeled Finally, we added 6 false positive ancestral syntenies, defined as sets of markers containing between 2 ≤ c ≤ d markers and spanning an interval of G of at most g markers (hence the gaps in this false positive contain g − c markers), where g is a parameter of the dataset. We considered the values g = 2, 5, 10. To simulate the fact that, in general, if ancestral syntenies are weighted according to their conservation in extant species, false positives have a lesser weight than true positives, we weighted false positives by 0.5 and true positives by 1.0. For each pair of parameters (d,g), we generated 10 datasets.
This generation method was designed to simulate moderately large datasets that resemble real datasets. For most of the 120 datasets, the generation of all MCS and MC1PS could be completed within three hours of computation, but for 13 of them (one for (d,g) = (3, 5), three for (4, 5), one for (5, 5), four for (3, 10), three for (4, 10) and two for (5, 10)) that were stopped if the computations were not completed after three hours. These unfinished datasets were discarded when computing the statistics described below.
Table 1 presents a summary of the number of MCS and MC1PS observed in all the completed datasets; for computations that had to be interrupted, similar results are observed from the partial information contained in the log files. We can first observe the number of MC1PS is much larger than the number of MCS. The second important observation is a general trend towards increasing the number of MCS when either d or g increases, which can be explained by the more intricate combinatorial structure of sets of ancestral syntenies. Indeed, for example, with d = 2 and g = 2, MCS are easy to find and count, as cycles in the corresponding bipartite graph are short and are relatively easy to find, even using brute-force approach. With larger values of g, cycles length increase and overlapping cycles appear more frequently, which results in more MCS. Although it seems natural that increasing the value of d results in more MCS understanding more precisely the impact of increasing d requires a better understanding of the combinatorial structure of MCS with matrices of degree larger than 2; see You (2009) for the case d = 3. Regarding MC1PS, we notice that increasing g results also in an increase of the number of MC1PS, but we also notice that when d attains the value 5, there seems to be a decrease of the number of MC1PS. A possible explanation is that, with such degree, constraints on sets of rows that have the C1P increase as a given row can now overlaps a large number of other rows, which reduces the number of MC1PS containing such rows. Generally, the impact of the degree on MC1PS deserves further theoretical or experimental investigations.
For each dataset, and each ancestral synteny, we also computed two statistics, the Conflicting Ratio (CR) and the MC1PS Ratio (C1PR). For every row, we also computed its MCS rank and MC1PS rank, defined as follows: the MCS (resp. MC1PS) rank of a row of M is its rank when rows are ordered by increasing CR (resp. increasing C1PR). Table 2 presents a summary of these statistics. We can notice that, in general, MCS seem to discriminate slightly better between false positives and true positives in terms of ratio: the average difference between the CR of a false positive and of a true positive is slightly larger than the average difference of the C1PR. The difference between the rankings is more strongly in favor of MCS: on average, a false positive is more likely to have a MCS rank close to the maximum rank than to have a low MC1PS rank. The conclusion we can draw from these average results is that the CR and MCS rank seem to better discriminate false positive from true positives.2 Considering the weights of the rows to weight MCS and C1PS does not change significantly this conclusion (results not shown).
FP_CR is the Conflicting Ratio for False Positives, TP_CR is for CR the True Positives, FP_MR is the MC1PS ratio for False Positives, and TP_MR is the MR for True Positives.
Confirming the conclusions from Table 2, we can observe in Figures 1 and 2, the following facts.
Percentage of conserved ancestral syntenies (y-axis) given a minimal C1PR (Threshold, x-axis), for both false positives (FP) and true positives (TP) for simulated datasets. Percentage of conserved ancestral syntenies (y-axis) given a maximal C1PR (Threshold, x-axis), for both false positives (FP) and true positives (TP) for simulated datasets. The conflicting ratio (CR) discriminates effectively between false positives and true positives. For example conserving all syntenies that have a CR at least 0.14 results in discarding 80% of FP while still keeping 83% of TP. The MC1PS ratio (C1PR) does not discriminate as effectively between false positives and true positives.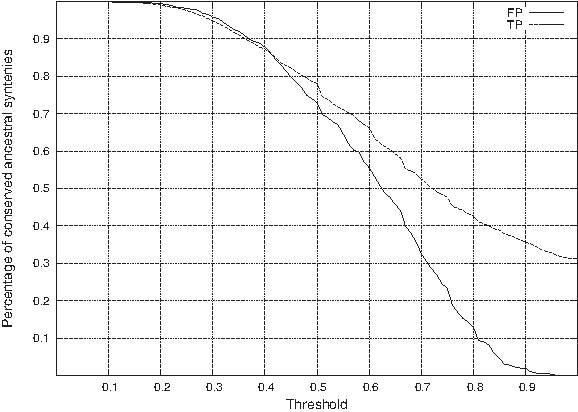
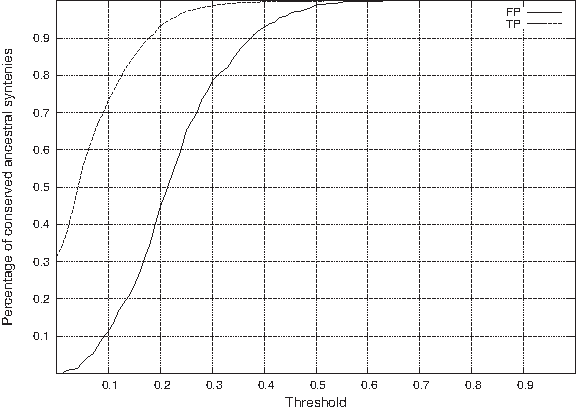
It is also interesting to note that only 31% of true positives ancestral syntenies do not belong to any MCS. This suggests that, at least with these simulated data, a significant number of ancestral syntenies do not need to be considered when trying to detect false positives (as we expect rows with CI equal to 0 to be true positives in general), but also that a very large part of true positive show some conflicting signal despite the low ratio of false positives. This shows that the extreme approach of discarding all rows belonging to at least one MCS, suggested in Bergeron et al. (2004), can result in discarding a very large number of true positives.
5.2. Application on yeasts real data
Next, we considered a real dataset, used to reconstruct an ancestral Saccharomycetaceae genome from non-duplicated yeasts genomes (S. kluyveri, K. thermotolerans, K. lactis, A. gossypii, and Z. rouxii), described in Chauve et al. (2010). These genomes are represented with 1420 markers,3 and a total of 3106 ancestral syntenies were computed, giving a binary matrix M with 3106 rows and 1420 columns. From this large matrix, five submatrices
Numbers marked by the symbol * correspond to partial results for interrupted computations.
We can observe that, unlike with simulated data, the number of MCS is larger than the number of MC1PS, a fact that is not related to the filtering of MC1PS. This could be explained by the fact that ancestral syntenies can be much larger here than in the simulated data (where they were of size at most 5) which can imply that a single false positive can create MCS with many different sets of true positive rows. The second important observation is that approximately 85% of all ancestral syntenies belong to all MC1PS (or at least, for M4 and M5, all generated MC1PS) and can then be considered as reliable. This fact addresses a question that is raised in Chauve et al. (2010) about the reliability of ancestors computed from ancestral syntenies, as it shows that most ancestral syntenies are reliable, at least from a combinatorial optimization point of view. To refine this observation, we show in Figure 3 that most ancestral syntenies have a high C1PR and a low CR.
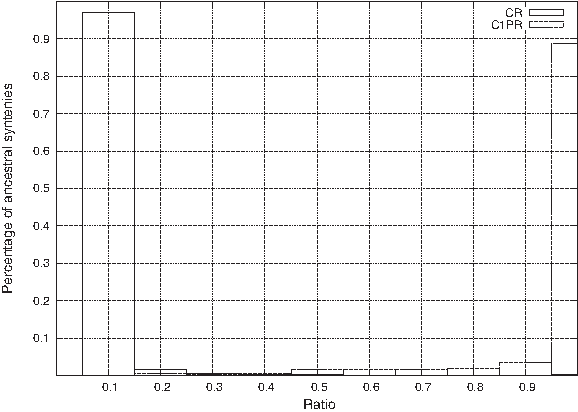
Percentage of conserved ancestral syntenies (y-axis) with a given CR (resp. C1PR). Each bar represents the percentage of ancestral syntenies whose CR (resp. C1PR) is in an interval of length 0.1.
Finally, we selected the subset of ancestral syntenies with a CR at most 0.5 and a C1PR at least 0.5. This subset of ancestral syntenies does not have the C1P, but only three ancestral syntenies need to be discarded to obtain a C1P matrix, that defines a set of 13 CARs. In comparison, the set of all ancestral syntenies from
6. Conclusion
This article describes preliminary theoretical and experimental results on Minimal Conflicting Sets and Maximal C1P Sets. In particular, we suggested that Tucker patterns are fundamental to understanding the combinatorics of MCS, and that the generation of all MCS is a hard problem, related to monotone boolean functions. From an experimental point of view it appears, at least on datasets of adjacencies, that MCS offer a better way to detect false positive ancestral syntenies than MC1PS. From a methodological point of view, it suggests that the joint generation framework provides a very general and flexible tool for handling the notion of minimal conflicting data in computational biology, as shown for example in Haus et al. (2008). This leaves several open problems to attack.
Detecting non-conflicting rows. The complexity of detecting rows of a matrix that do not belong to any MCS when rows can have an arbitrary number of entries 1 is still open. Solving this problem probably requires a better understanding of the combinatorial structure of MCS and Tucker patterns. Tucker patterns have also be considered in Dom (2008), where polynomial time algorithms are given to compute a Tucker pattern of a given type for a matrix that does not have the C1P. Even if these algorithms can not obviously be modified to decide if a given row belongs to a given Tucker pattern, they provide useful insight on Tucker patterns.
It follows from the dual structure of monotone boolean functions that the question of whether a row belongs to any MCS is equivalent to the question of whether it belongs to any MC1PS. Indeed, for an arbitrary oracle-given function, testing if a variable appears in any MTC is as difficult as deciding if a list of MTC is complete. Consider an oracle-given f and a list of its MTC which define a (possibly different) function f′. We can build a new oracle function g with an additional variable x0, such that g(x0,x) = 1 if and only if x0 = 0 and f′(x) = 1 or x0 = 1 and f (x) = 1.
Generating all MCS and MC1PS. Right now, this can be approached using the joint generation method, but the number of MCS and MC1PS makes this approach time consuming for large matrices. A natural way to deal with such problem would be to generate at random and uniformly MCS and MC1PS. For MCS, this problem is at least as hard as generating random cycles of a graph, which is known to be a hard problem (Jerrum et al., 1986). We are not aware of any work on the random generation of MC1PS.
An alternative to random generation would be to abort the joint generation after it generates a large number of MCS and MC1PS, but the quality of the approximation of the MCS ratio and MC1PS ratio so obtained would not be guaranteed. Another approach for the generation of all MCS is based on the remark that, for adjacencies, it can be reduced to generating all claws and cycles of the graph GM. Generating all cycles of a graph can be done in time that is polynomial in the number of cycles, using backtracking (Read and Tarjan, 1975). It is then tempting to use this approach in conjunction with dynamic partition refinement (Habib et al., 2000) for example or the graph-theoretical properties of Tucker patterns described in Dom (2008).
Combinatorial characterization of false positive ancestral syntenies. It is interesting to remark that, with matrices of degree 2, most false positives can be identified in a simple way. True positive rows define a set of paths in the graph GM, representing ancestral genome segments, while false positive rows {i,j}, unless i or j is an extremity of such a path (in which case it does not exhibit any combinatorial sign of being a false positive), both the vertices i and j belong to a claw in the graph GM. And it is easy to detect all edges in this graph with both ends belonging to a claw. In order to extend this approach to more general datasets, where Δ(M) > 2, it would be helpful to better understand the impact of adding a false positive row in M. The most promising approach would be to start from the partition refinement (Habib et al., 2000) obtained from all true positive rows and form a better understanding of the combinatorial structure of connected components of the overlap graph that do not have the C1P.
Computation speed. On large datasets, especially with matrices with an arbitrary number of entries 1 per row, some connected components of the overlap graph can be very large, see the data in Chauve et al. (2010), for example. In order to speed up the computations, algorithmic design and engineering developments are required, both in the joint generation algorithm and in the problem of testing the C1P for matrices after rows are added or removed.
7. Appendix: The Five Tucker Patterns
Footnotes
Acknowledgments
Cedric Chauve and Tamon Stephen were partially supported by NSERC Discovery Grants. Utz-Uwe Haus was supported by the Magdeburg Center for Systems Biology, funded by a FORSYS grant of the German Ministry of Education and Research. Vivija P. You was partially supported by The Department of Mathematics of Simon Fraser University. We would also like to acknowledge the support of the IRMACS Centre at SFU. A preliminary version of this paper appeared as Chauve et al. (2010).
Disclosure Statement
No competing financial interests exist.
1
2
The opposite conclusion was stated in the preliminary version of this article (Chauve et al., 2010), due to an experimental error.
3
The 1420 markers represent 710 synteny blocks, each block being split into two markers corresponding to its two extremities and represented by an ancestral synteny of size 2 containing these two markers.
4
Each matrix corresponds to an R-node of the PQR-tree associated to M; see Chauve and Tannier (2008;) and McConnell (![]() ) for details on R-nodes in PQR-trees.
) for details on R-nodes in PQR-trees.
5
For each matrix, we filtered the set of MC1PS to discard any set of rows that does not contain all ancestral syntenies corresponding to the synteny blocks represented by the columns of this matrix.