
Abstract
Abstract
Studies conducted during the last decade unexpectedly revealed several new biological functions of RNA molecules. The involvement of RNA in many complex processes requires highly effective systems controlling its accumulation. In this context, the mechanisms of degradation appear as one of the most important factors influencing RNA activity. Here, we present our first attempt to describe the RNA degradation process using bioinformatics methods. Based on the obtained data, we propose a formulation of a new problem, called RNA Partial Degradation Problem (RNA PDP) and the algorithm that is capable of reconstructing an RNA molecule using the results of biochemical analysis of its degradation. In addition, we present the results of biochemical experiments and computational tests.
1. Introduction
The level of RNA accumulation is regulated by maintaining balance between transcription and degradation pathways. Consequently, the stability of RNA molecules is one of the major factors shaping the composition of cellular transcriptome. In eukaryotic cells, the degradation of mRNA is initiated by the removal of a poly(A) tail by deadenylases. Next, the RNA molecule is digested by 3′-5′ exonucleases in exosome. In addition, DCP2 protein-mediated decapping causes mRNA to be a proper substrate for 5′-3′ exoribonucleases (Eulalio et al., 2007). Many of the enzymes and cofactors involved in RNA degradation participate also in RNA processing or maturation (Houseley et al., 2009). Although all RNA molecules present in a given cell are exposed to the same RNA degradation machinery, some of them exist and function for a longer time than others. The half-life of mRNAs can range from hours to seconds (Lorentzen et al., 2006). Unfortunately, at present, our knowledge about different factors affecting RNA degradation is very limited. Accordingly, one cannot predict the stability of individual RNAs. Some short-living mRNAs have been shown to carry adenosine and uracil rich cis-acting elements (ARE) usually located in their 3′ UTRs. These molecules undergo rapid degradation by the ARE-mediated mRNA decay (AMD) (Houseley et al., 2009). To avoid errors in RNA biogenesis or function, aberrant or nonfunctional RNAs are preferentially degraded by numerous quality control systems. One of them, nonsense-mediated decay (NMD), enables degradation of mRNAs carrying nonsense mutations (e.g., premature translation termination codon). Quality control systems are also involved in the repression of function or in degradation of viral and parasitic RNAs (Doma et al., 2007).
In contrast with mRNA, the degradation pathways of relatively stable molecules like tRNA and rRNA, accounting for more than 90% of total cellular RNA, have been poorly characterized. In bacteria, the degradation of stable RNAs is usually associated with starvation (Deutscher, 2003). Recent reports have shown that rRNA and tRNAs can undergo cleavage in response to oxidative stress or under developmental regulation (Thompson et al., 2008). Now, the most important question concerns biological function of the cleavage products. There is some evidence suggesting that they may function as translation inhibitors or signaling molecules (Kierzek, 1992; Zhang et al., 2009).
Better understanding of RNA degradation is indispensable, not only for broadening our knowledge on the physiological functions of RNA molecules, but it is also necessary if one wishes to modulate a half-life of any RNA molecule and in this way regulate its biological activity (e.g., by influencing the stability of mRNA one can affect the level of protein accumulation). A large number of factors can affect RNA stability. Generally, they can be divided into two groups: (i) factors acting in trans—all cellular factors; and (ii) factors acting in cis—RNA primary, secondary, and tertiary structure.
Although many RNAs are similarly accessible by the degradation machinery, some molecules are more stable than the others. It suggests that the primary, secondary, and tertiary structures of RNA molecules significantly affect their degradation.
Unfortunately, currently available experimental methods do not allow for studying all aspects of this process. Experiments in model in vitro systems, however, can be performed in well-defined conditions.
In this article, we present our first attempt to describe the RNA degradation process using bioinformatics methods. Our studies were focused on cis acting factors affecting RNA stability and involved two artificial RNA molecules. Both of them were designed in such a way that they contained well-defined unstable regions (Bibillo et al., 1999, 2000; Kierzek, 2001). The undertaken biochemical and bioinformatics analyses confirmed the predicted pattern of RNA degradation. Based on the obtained data, we proposed a formulation of a new problem, called RNA Partial Degradation Problem (RNA PDP) and the algorithm that is capable of reconstructing RNA molecule using results of biochemical analysis of its degradation. By solving the RNA PDP problem, one can determine the location of cleavage sites within analyzed RNA molecule having as an input the lengths of fragments being the degradation products. As a result, one can identify the unstable regions of RNA molecules which are the most susceptible to the degradation.
The organization of the article is as follows: Section 2 discusses the combinatorial model and gives the strong NP-completeness proof of the decision version of RNA PDP problem, which is equivalent to a non-existence of a polynomial-time exact algorithm for the problem in question (Garey et al., 1979). sSection 3 presents the exact algorithm for RNA PDP based on the branch-and-cut idea. In Section 4, the results of the biochemical experiment and computational tests are given. Section 5 points out the directions for further research.
2. Problem Formulation and Complexity
To analyze the degradation process dependent on cis acting elements (RNA structure), the following two types of experiments have been carried out: (i) involving multi-labeled RNA-32P labeled nucleotides were randomly introduced along the RNA molecule; and (ii) involving single labeled RNA-32P was introduced at the 5′ terminus of the tested RNA. The first type of experiment was carried out to visualize all fragments generated during degradation. In this case the information of exact position of the fragments within analyzed molecule is missing. The second type of experiment was carried out to visualize only these degradation products which contained labeled 5′ end. In this case, their location within tested molecule is known. Thus, we see that two collections of fragments are created. Each fragment is represented by its length. For further mathematical considerations we will use the lengths as specified parameters of fragments. Let
With respect to the above description of the problem, we propose its mathematical model that will serve as a background for the complexity analysis and for the construction of the algorithm that solves the problem.
The mathematical formulation of the RNA Partial Degradation Problem is presented below. P1 stands for the set of primary cleavage sites in the solution and P2 for the set of secondary ones.
Problem 1.
RNA Partial Degradation Problem—decision version (ΠRNAPDP).
Instance
Multiset
Question
Do there exist sets P1 and P2 such that:
Here, the general version of the problem is assumed, where missing elements, i.e., false negatives, in D and Z are allowed. ΠRNAPDP is the decision version of the optimization problem, in which the total number of false negatives is minimized. In the formulation, D′ and Z′ are the complete counterparts of the sets, and C is the false negatives bound. Assigning C to 0 we get the ideal problem with no false negatives allowed.
In this section, we prove strong NP-completeness of problem ΠRNAPDP by a pseudo-polynomial transformation from problem Numerical Matching With Target Sums, cited below (of course, the strong NP-completness of ΠRNAPDP implies strong NP-hardness of the search version of the RNA PDP). This guides a choice of the proper solution strategy for the latter: either an exact exhaustive search algorithm or a polynomial-time heuristic strategy, not guaranteed to find an exact solution.
Problem 2.
Numerical Matching With Target Sums (ΠNM)—decision version.
Instance
Disjoint sets X and Y, each containing q elements, sizes s(xi) and s(yi) for every
Question
Can X ∪ Y be partitioned into q disjoint sets
This problem is strongly NP-complete (Garey et al., 1979).
The transformation proposed below in its first step (modification of ΠNM) uses some ideas of our former proof for a distinct problem of the Simplified Partial Digest (SPDP) of DNA molecules (Blazewicz et al., 2005; cf. also Waterman, 1995; Blazewicz et al., 2001, 2007). For the moment, the dependence between somewhat similar problems: RNA PDP and PDP as well as SPDP, is not yet found out, being an interesting question for further studies.
The first stage of proving strong NP-completeness of problem ΠRNAPDP consists in a simple modification of problem ΠNM. We are interested in a variant with the ranges of variables shifted by some added values. The modified ranges will have several properties, in particular, they will be disjoint. Initially, the ranges of values of variables s(xi), s(yi), and ui, 1 ≤ i ≤ q, can be written as follows:
where the variables with index L mean the smallest values and the ones with index R mean the largest values in the respective collections. These ranges are arbitrary; however, we assume here that they satisfy a few obvious conditions:
The above assumptions do not change the computational complexity of problem ΠNM—if one of them is not satisfied, the problem becomes easy because the answer is obviously “no.”
We modify the initial ranges of the variables in the following way (where “←” assigns the right-hand side value to the left-hand side variable):
The new ranges have the following form (1 ≤ i ≤ q):
They are visualized in Figure 1.
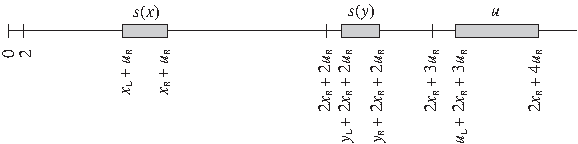
The ranges of values of variables s(xi), s(yi), and ui, 1 ≤ i ≤ q, after the modification.
As a result we have:
Lemma 1
(Blazewicz et al., 2005). Problem ΠNM and its version with the ranges of variable values shifted as above, are equivalent.
The variables from problem ΠNM increased by the proposed values have the following useful properties.
Lemma 2
(Blazewicz et al., 2005). None of the modified ui, 1 ≤ i ≤ q, can be equal to some s(xj) or to a sum of any s(xj) and s(xk).
Lemma 3
(Blazewicz et al., 2005). None of the modified ui, 1 ≤ i ≤ q, can be equal to some s(yj) or to a sum of any s(yj) and s(yk).
Now, we can define the transformation from problem ΠNM to problem ΠRNAPDP, which is given below.
The transformation
Given an instance of problem ΠNM, the corresponding instance of ΠRNAPDP is constructed as follows.
(1) Shift the ranges of numbers in problem ΠNM as specified above, i.e.,
(2) Create set (3) Assign values to L and C: L = max(Z), C = 0.
Lemma 4.
The proposed transformation can be computed in time bounded by a polynomial in the length of the instance of ΠNM (LenNM) and the maximal number appearing in this instance (MaxNM).
Proof.
LenNM is O(q⌈log MaxNM ⌉ ). In the first step of the transformation we make O(LenNM) operations. New values of the variables do not change LenNM and MaxNM substantially: q is not changed, new MaxNM is up to 6 times larger than previously. Filling set Z requires O(LenNM2) operations. Filling multiset D requires O(LenNM2⌈log LenNM ⌉ ) operations. Step (3) is O(LenNM⌈log LenNM ⌉ ). Taking the above functions together, we have O(LenNM2⌈log LenNM ⌉ ) as the complexity of the proposed transformation. ▪
Because the instance size does not decrease exponentially and the greatest number in the instance does not increase exponentially, this transformation is pseudo-polynomial. We can now prove the following main theorem.
Theorem 1.
RNA Partial Degradation Problem (ΠRNAPDP) is strongly NP-complete.
Proof.
Lemma 4 proves that the proposed transformation is pseudo-polynomial. It remains to prove that the transformation is correct, i.e. that for every instance I of problem ΠNM,
Let us assume, that an instance of problem ΠNM gives a positive answer. It means, that there is a partition of X ∪ Y such that every disjoint subset Wi, 1 ≤ i ≤ q, contains one xj and one yl, and s(xj) + s(yl) = ui, for some
Now let us assume, that an instance of problem ΠRNAPDP (after the transformation) has answer “yes.” In such a case, we have sets P1 and P2 satisfying all 9 constraints from the definition of ΠRNAPDP. We know, that constructed sets Z and D do not contain false negatives (because C = 0), so Z = Z′ and D = D′. Set Z \ {L} indicates q cleavage sites of a molecule of length L, which define segments of lengths ui, 0 ≤ i ≤ q. The instance of ΠRNAPDP contains in multiset D 2q values less than any ui, i > 0, among them q values from the range of s(x) and q values from the range of s(y) (Fig. 1). The addition of any of these “segments” to the set of cleavage sites produced by Z \ {L} would cause the addition of at least one new cleavage site. We know, that they can be only secondary cleavage sites of primary segments not beginning in 0, because otherwise they would increase the false negatives pool (constraints 6–7). The positive answer of problem ΠRNAPDP forces such pairing of the segments that they cover all (except the one beginning in 0) primary segments of the lowest level, i.e., the segments of lengths ui, 1 ≤ i ≤ q. These segments cannot cover primary segments of a higher level (i.e., primary segments containing other primary segments), because no two lengths from the ranges of s(x) and s(y) sum up to such big value. The properties of the shifted ranges of s(x) and s(y) guarantee, as the only possible assignment, always one element belonging to s(x) and one to s(y) in every segment ui, 1 ≤ i ≤ q (see Lemmas 2–3). The solution of problem ΠNM is the set of pairs Wi = {xj, yl} such that s(xj) and s(yl) fill up the ith segment of length ui, 1 ≤ i ≤ q. In such a case P1 = Z \ {L}, P2 equals to all newly added cleavage sites and all not yet used elements of D cover (following the construction) all interpoint distances between pairs of values from P1 ∪ {0, L}. This ends the proof. ▪
The proposed transformation of an instance of problem ΠNM to an instance of problem ΠRNAPDP is illustrated by the following example.
Example 1.
Let the example instance of problem ΠNM be:
After shifting the ranges of the variables we get the values:
The construction of the instance of problem ΠRNAPDP ends with the following result:
The feasible solution for the instance after the transformation (i.e. of problem ΠRNAPDP) is shown in Figure 2, with P1 = {1, 45, 91} and P2 = {12, 57, 105}.
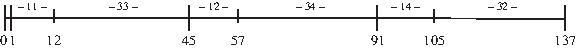
A solution of problem ΠRNAPDP for the example instance.
This solution can be easily translated to a feasible solution of problem ΠNM:
Corollary 1.
RNA Partial Degradation Problem in its decision version without any false negatives allowed (i.e., with C = 0) is strongly NP-complete.
Corollary 2.
RNA Partial Degradation Problem in its search version is strongly NP-hard.
Following this result, we will propose in the next section an exhaustive search algorithm which finds first cleavage sites, however, at the expense of the computational time.
3. The Exact Algorithm
In this section, we introduce the branch-and-cut algorithm that works for the case of RNA PDP problem (search version) with false negatives (i.e., missing fragments in D and Z). Its aim is to find the coordinates of primary and secondary cleavage sites in a given RNA molecule, taking into account false negatives. The proposed algorithm builds and searches through the solution tree with leaves corresponding to elements of the solution space of the problem. The algorithm is implemented in C programming language and runs in Unix environment.
The algorithm takes as an input the data containing fragment lengths obtained via the biochemical experiments, i.e., multiset D of k positive integers and set Z of n positive integers, and also maximum permissible number of false negatives (maxErr = C). In addition, the following structures are defined: multiset R of all pairwise distances between points in P1 ∪ {0, L}, where each distance is described by a pair of points, and sorted (in non-increasing order) list B of all secondary fragment lengths. These structures do not contain any element at the beginning of the algorithm. The output of the algorithm is set P1 ∪ P2 which will contain reconstructed primary and secondary cleavage sites in RNA molecule.
The aim of the preliminary step of the algorithm is to check whether Z is consistent with D. If not, D is increased by elements of Z missing there. The output of this step is an updated multiset D and the current number of corrected false negatives (currErr). If currErr ≤ maxErr is not satisfied, the solution does not exist.
The main algorithm consists of two parts. The first part was designed to find the coordinates of primary cleavage sites including false negatives (see Algorithms 1 and 2). In this part, P1 is recognized. If a given cleavage site cannot be assigned due to high currErr, the current branch is cut off and the algorithm backtracks. First, each of the elements of Z is checked whether it can be classified as a primary cleavage site as shown in pseudo-code below (Algorithm 1). The number of immediate successors of the node is equal to two, because the element of Z can be added to set P1 as a primary cleavage site or denoted as a secondary cleavage site and added to list B. Next, the algorithm attempts to reconstruct the missing primary cleavage sites basing on D in a very similar way as for Z (Algorithm 2). Each element of D \ {L} which is not equal to any of the distances between reconstructed so far primary cleavage sites in P1 including ends of the molecule and does not belong to list B is considered as a potential primary cleavage site. Every such element can be placed either on the left or right side of the RNA molecule and inserted into P1 or added to the list of secondary fragments B. Finally, P1 will contain reconstructed primary cleavage sites. The main procedures of the first part, given in pseudo-code, are shown below as Algorithms 1 and 2, respectively. The presented pseudo-code works for the case of ideal data and gives an overview of the algorithm. The notation Δ(zmax, P1 ∪ {0, L}) denotes the multiset of distances between point zmax and all points in set P1 ∪ {0, L}. The procedure stops when the stopping criterion is satisfied, defined as follows. If set Z \ {L} becomes empty, then the procedure ends with reconstructed primary cleavage sites in P1.
Reconstruction of primary cleavage sites basing on set Z (the case of data without false negatives)
Reconstruction of primary cleavage sites basing on set D (the case of data without false negatives)
As a result of the first part of the algorithm, the solution set P1 will contain all reconstructed primary cleavage sites.
The second part of the algorithm was designed to reconstruct the coordinates of the secondary cleavage sites including false negatives. In this part it is checked whether there exists the pair of elements in list B of secondary fragment lengths, which sum is equal to one of the primary fragment lengths. The secondary cleavage site can also be reconstructed when one of the elements of the pair is missing, but only if currErr < maxErr. Additionally, only one of the elements in the pair and only within primary fragment starting with the first nucleotide of the RNA of length L, can correspond to a secondary cleavage site obtained during experiment with labeled 5′ termini of the RNA molecule. If a pair could not be found, then the current branch is cut off and the algorithm backtracks. The number of immediate successors of the node is equal to a number of pairs which can be composed of the first (i.e., the longest) element of the current list B and any other element of B (including possible missing fragment if currErr < maxErr) in such way that these two elements sum up to a length of a primary fragment within current P1. The main procedure of the second part consists of the presented steps in pseudo-code shown as Algorithm 3. The presented pseudo-code works for the case of ideal data and gives an overview of the algorithm. The procedure stops when the stopping criterion is satisfied, defined as follows. If list B becomes empty then the procedure ends with reconstructed secondary cleavage sites in P2. If a solution is not found because of the higher number of false negatives than assumed, then the algorithm backtracks to Part I. A new reconstruction of primary cleavage sites is searched for.
Reconstruction of secondary cleavage sites (the case of data without false negatives)
The whole algorithm stops after finding a first feasible solution of the considered problem. The algorithm was also extended to cover the case of errorneous data.
The example below illustrates the case with false negatives.
Example 2 presents in detail the action of the proposed algorithm.
Example 2.
Let our instance of the considered problem ΠRNAPDP be: D = {19, 21, 23, 24, 25, 26, 43, 45, 51, 66}, Z = {24, 43, 45, 66}, L = 66 and C = maxErr = 5 (because the fragment of length 15 is missing in both Z and D, the corresponding false negative is counted twice). Exemplary reconstruction of cleavage sites can be as follows. As a result of the reconstruction of primary cleavage sites from Z in Part I of the algorithm, we will obtain: P1 = {43, 45}, B = {24} and currErr = 1. Next, elements of D \ {L} that do not belong to set P1 ∪ B and are not equal to any distances between elements of P1 and endes of the molecule are selected as potential primary cleavage sites. These elements are: 19, 25, 26, 51. The reconstruction of the primary cleavage sites from D \ {L} will result in: P1 = {15, 43, 45}, B = {26, 25, 24, 19} and currErr = 5. In Part II of the algorithm, the secondary cleavage sites are reconstructed. The solution set will be P1 ∪ P2 = {15, 24, 41, 43, 45}.
4. Experimental Results
In this section, results of the biochemical experiments and the tests of the algorithm solving the RNA PDP problem in the case of erroneous data are presented. The algorithm has been tested on a PC with Pentium(R) 4, 2.40-GHz processor, and 1-GB RAM in Unix environment. As a testing set, a group of experimental and randomly generated data were prepared. The biochemical analysis of RNA degradation was conducted using two artificial molecules RNA-A108 and RNA-B66. The secondary structures of both molecules were designed on the base of earlier described rules of nonenzymatic degradation (Kierzek, 1992, 2001; Bibillo et al., 1999, 2000) and the program mfold (Fig. 3).
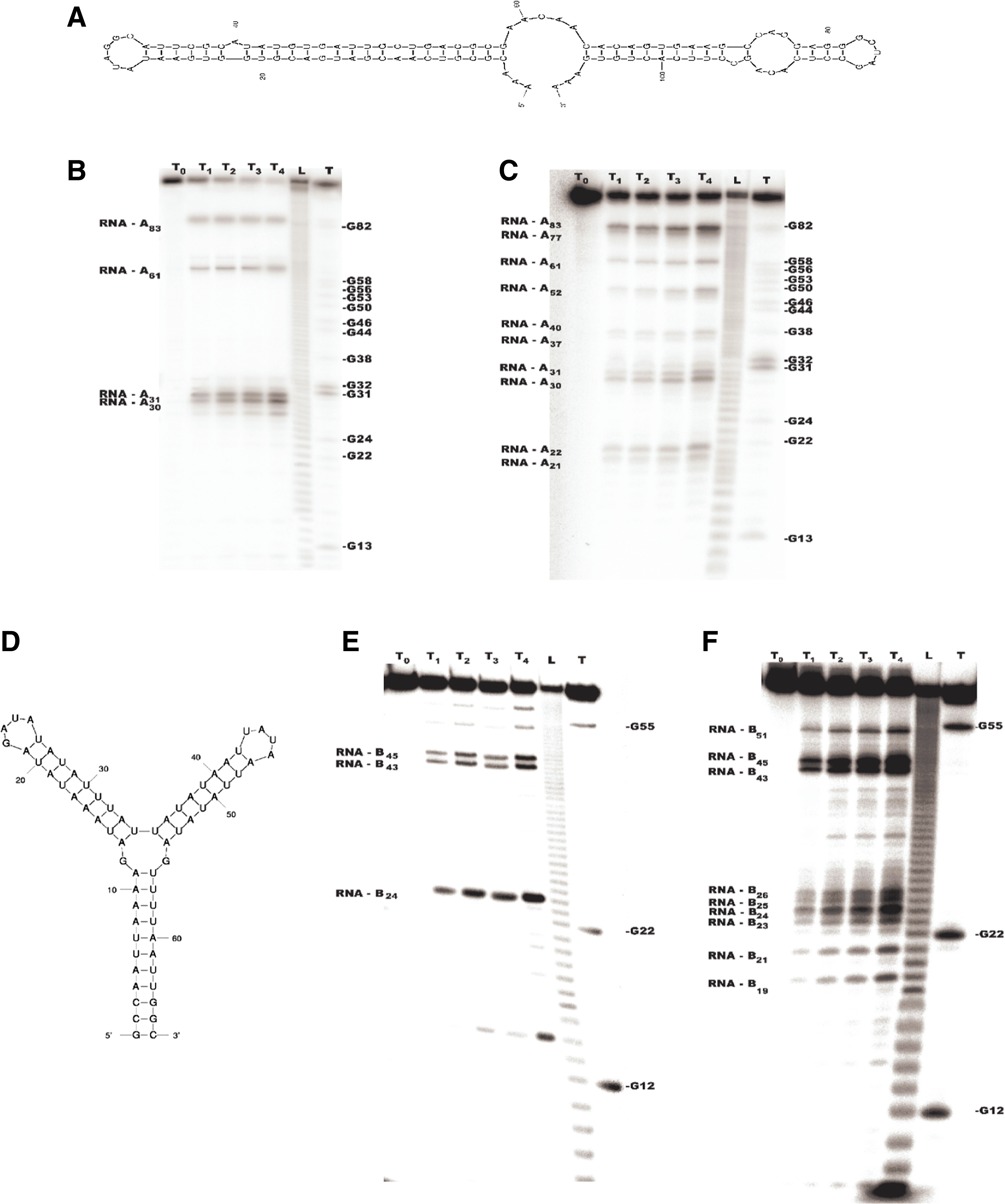
Secondary structure models of two artificial RNA molecules: RNA-A108
RNA-A108 and RNA-B66 molecules were obtained and labeled as described in the literature (Dutkiewicz et al., 2005). Briefly, both RNAs were synthesized by in vitro transcription involving chemically synthesized DNA as templates. The resultant transcripts were labeled in two different ways; either a single 32P atom was incorporated at their 5′ terminus (single-labeled RNA) or numerous 32P atoms were introduced along the molecules (multi-labeled RNA). To prepare single-labeled RNA, a transcription product was purified and subjected to reaction with [γ-32P]ATP and T4 polinucleotide kinase. To obtain multi-labeled RNA, [α-32P]UTP was added to the in vitro transcription reaction mixture. All reactions were conducted with the MEGAshortscript™ kit (Ambion), and resultant transcripts were purified as described earlier (Dutkiewicz et al., 2005).
RNA degradation experiments were carried out according to procedure proposed for studying nonenzymatic RNA hydrolysis (Kierzek, 1992, 2001; Bibillo et al., 1999, 2000). Reaction mixture containing 0.1 pmol of labeled RNA, 50 mM Tris-HCl (pH 7.5), 2 mM EDTA, 1 mM spermidine, 50 mM NaCl, and 0.1% PVP solution was incubated at 37∘C for 1, 2, 4, and 6 h. The reaction products were separated in a 16% denaturating polyacrylamide gel and visualized by autoradiography. The locations of cleavage sites were determined by applying standard biochemical methods, by the comparison of the length of degradation products with the length of the RNA markers. The RNA markers were generated using typical procedures. In order to obtain the first set of RNA markers, the examined RNA was single-labeled at the 5′ end and subjected to random hydrolysis. As a result, we obtained so-called ladder (marked with L in Fig. 3), i.e., a mixture of labeled RNA which lengths ranged between a single nucleotide and a full length RNA. To produce the second set of markers, the examined RNA was single-labeled at the 5′ end and subjected to cleavage by RNase T1. It cuts RNA after G, thus we obtained a mixture of RNAs labeled at the 5′ end and having G at the 3′ end (marked with T1 in Fig. 3). The results of the experiment are shown in Figure 3. Their analysis revealed that primary and secondary cleavages occurred during the first hour of incubation. For this reason we considered in further computational tests only those fragments that arose at T1. During the degradation of single-labeled RNA-A108, the following 5′ radiolabeled fragments were generated: 30, 31, 61, 83, and 108 nucleotides long. The degradation of single-labeled RNA-B66 led to the formation of 4 radiolabeled fragments: 24, 43, 45, and 66 nucleotides long. In the second experiment with multi-labeled RNA-A108 11 fragments were detected: 21, 22, 30, 31, 37, 40, 52, 61, 77, 83, and 108. During similar experiment involving RNA-B66 we observed the formation of 10 products: 19, 21, 23, 24, 25, 26, 43, 45, 51, and 66.
The above data were used to perform computational tests. Although all of the instances gathered in the biochemical experiments had a high number of missing primary and secondary fragments, the algorithm was able to find the correct solution. It was able to determine the following primary cleavage sites in a case of RNA-A108: 31, 83; and RNA-B66: 15, 43, 45. The reconstructed secondary cleavage sites were as for RNA-A108: 30, 52, 61, 71; and for RNA-B66: 24, 41. Analyzing the obtained results, we noticed that the algorithm performs very fast for the real data. Table 1 summarizes running time results for the branch-and-cut algorithm tested on the above instances.
Since the real instances were solved very quickly, random instances with a higher number of primary and secondary cleavage sites as well as a defined number of false negatives were generated. The random erroneous data were obtained by generating sequences of random numbers, from a uniform distribution over interval [1, 2500]. First the primary cleavage sites were generated. The test data have been divided into four sets where secondary cleavage sites have occurred in, respectively, 25%, 50%, 75%, and 100% of primary fragments, the number of the latter being equal to
Secondary cleavage sites occur in 25% of all primary fragments.
Secondary cleavage sites occur in 50% of all primary fragments.
Secondary cleavage sites occur in 75% of all primary fragments.
Secondary cleavage sites occur in 100% of all primary fragments.
Analyzing the obtained results, we noticed that the algorithm performs quite efficiently and fast, despite its computational complexity, which equals O(r22 o ), where r is the number of primary cleavage sites and o stands for the number of secondary fragment lengths (a pair of secondary fragment lengths corresponds to one secondary cleavage site). The algorithm has been able to find a correct solution in all the considered examples.
5. Conclusion
In this article, the new RNA PDP has been formulated and solved. The mathematical formulation of the problem has been shown, and its complexity has been analyzed. Since the problem has been proved to be strongly NP-complete, a branch-and-cut algorithm has been proposed. This algorithm gives very good results for erroneous data, especially for the real data obtained from the RNA degradation, and may be very useful in practice as a tool that facilitates the analysis of the output of the biochemical experiment.
In the RNA PDP, we have assumed that a primary fragment is cleaved at most once. Certainly, there could be in general several secondary cleavage sites in between when allowing for long enough RNA incubation. In fact, very long incubation should lead to complete RNA degradation into single nucleotides. However, the problem of the reconstruction of the whole history of an individual RNA from the full length molecule to single nucleotides is too complex. Thus, we have decided to focus our analysis on the preferred cleavage sites and this way simplify the general problem to make easier the reconstruction of the original molecule. The proposed formulation of the simplified problem is enough general to model the degradation of the analyzed RNA and at the same time is still tractable for the reader.
As a continuation of the research reported in this article, one may consider the analysis of not only secondary but also further products of the spontaneous RNA degradation that occur after several hours. Consequently, we should expect that adding next-order cleavage sites into the search space will complicate the formulation of the problem and the searching algorithm.
Footnotes
Acknowledgments
This research was supported by the Polish Ministry of Science (grants N-N519-314635 to M.K. and PBZ-MNiI-2/1/2005 to M.F.).
Disclosure Statement
No competing financial interests exist.