
Abstract
Abstract
The automatic classification of protein sequences into families is of great help for the functional prediction and annotation of new proteins. In this article, we present a method called Irredundant Class that address the remote homology detection problem. The best performing methods that solve this problem are string kernels, that compute a similarity function between pairs of proteins based on their subsequence composition. We provide evidence that almost all string kernels are based on patterns that are not independent, and therefore the associated similarity scores are obtained using a set of redundant features, overestimating the similarity between the proteins. To specifically address this issue, we introduce the class of irredundant common patterns. Loosely speaking, the set of irredundant common patterns is the smallest class of independent patterns that can describe all common patterns in a pair of sequences. We present a classification method based on the statistics of these patterns, named Irredundant Class. Results on benchmark data show that the Irredundant Class outperforms most of the string kernels previously proposed, and it achieves results as good as the current state-of-the-art method Local Alignment, but using the same pairwise information only once.
1. Introduction
A number of methods have been proposed for the protein sequence classification. Historically this problem has been studied, for quite some times, in the field of text documents classification. Unfortunately most of the proposed approaches, developed for a different kind of strings, fail when applied to biological sequences. The main reasons of this failure are the different nature of biological sequences, particularly rich of regularities known as patterns that are difficult to digest for a general purpose application.
The main distinction is between generative methods against discriminative methods. The former class includes methods such as protein family profiles (Gribskov et al., 1987), hidden Markov models (HMMs) (Krogh et al., 1994; Baldi et al., 1994; Karplus et al., 1998), and PSI-BLAST (Altschul et al., 1997). These methods tend to derive a model for a set of proteins and then check whether a candidate protein fits the model or not. Unlike generative methods, discriminative methods (such as Jaakkola et al., 1999; Liao and Noble, 2003; Leslie et al., 2002, 2004; Saigo et al., 2004; Hou et al., 2003; Rangwala and Karypis, 2005; Kuang et al., 2005) focus on finding which sequences can describe a set of proteins despite of another set.
Recent results (Liao and Noble, 2003; Leslie et al., 2004) suggest that the best-performing methods are discriminative string kernels. These methods use kernel functions based on common patterns of pairs of protein sequences to train a support vector machine (SVM) (Vapnik, 1998; Cristianini and Shawe-Taylor, 2000). The string kernel extracts information from sequences and computes either a feature vector for each sequence or directly a kernel matrix with scores between pairs of sequences.
The first string kernel, called Fisher's kernel (Jaakkola et al., 1999), uses an HMM to provide the necessary means of converting proteins into fixed-length vectors. The vector summarizes how different the given sequence is from a typical member of the given protein family. In the Pairwise kernel (Liao and Noble, 2003), the feature vector corresponding to a protein sequence is formed by all E-values, given by the Smith-Waterman algorithm (Smith and Waterman, 1981), computed on the sequence analyzed and each of the training sequences of a particular experiment.
The Spectrum and the Mismatch kernels (Leslie et al., 2004, 2002) use as protein features the set of all possible substrings of amino acids of fixed length (k-mers). If two sequences contain many of the same k-length contiguous subsequences, their inner product under the k-Spectrum kernel will be large. Equivalently, the Mismatch kernel computes a large inner product between two sequences if these sequences contain many k-length contiguous subsequences that differ by at most e mismatches.
The Local Alignment method (Saigo et al., 2004) mimics the behavior of the Smith-Waterman algorithm to build a family of valid kernels. Following the work of Haussler (1999), they defined a kernel that mimics the detection of all local alignments between two sequences by convolving simpler kernels. In the Word Correlation Matrices method (Lingner and Meinicke, 2008), the kernel is defined by average pairwise word similarity between two sequences. The consequent analysis of discriminative words allows also for the identification of characteristic regions in biological sequences.
Other methods, such as the I-Sites (Hou et al., 2003), encode into feature vectors information related both to three-dimensional structure properties and sequence similarities of proteins. Or, like the eMOTIF-database method (Ben-Hur and Brutlag, 2003), they define a kernel function in terms of occurrences of sequence motifs previously stored in databases, and tipically extracted using popular algorithms on reference sequences. In particular, the Profile-based Mismatch methods (Kuang et al., 2005) use probabilistic profiles, such as those produced by the PSI-BLAST algorithm, to define kernels based on position-dependent mutation neighborhoods of k-mers with mismatches (in a similar way to the original Mismatch kernel). In contrast, the Profile-based Direct methods (Rangwala and Karypis, 2005) build kernel functions combining sequence profiles obtained with different approaches for determining the similarity between pairs of protein sequences. Note that the latter methods make an extensive use of hyperparameters, increasing the risk of overfitting.
We selected for comparison with our method some of the algorithms presented above, in particular those with state-of-the-art performance on the classification of proteins and which seem somehow more reliable: Fisher, Pairwise, Spectrum, Mismatch, Local Alignment (version “eig”), and Word Correlation Matrices. In general, all pattern-based methods operate two distinct steps: first extract and process common patterns from pair of sequences, then use this set of patterns as features to build an automatic classification tool based on SVMs, and so does the method proposed here. As we will show in the next sections, almost all string kernels are based on patterns that are not independent, namely patterns with occurrences that are related each other. Any score built using a set of related patterns is in practice based on redundant features, and therefore it can potentially overestimate the similarity score.
In this article, we want to stress the idea that if the learning process has to deal with a set of redundant features, this might mislead the classification. The goal is somehow similar to the feature selection problem, but in the case of pattern-based methods for classification contexts, the class of irredundant common patterns is specifically designed to address the issue of a repeated information. Our conjecture is that a set of irredundant common patterns, and consequently a set of independent features, can facilitate the automatic learning and classification of sequences.
2. Methods
2.1. The irredundant class
The method is based on the extraction and filtering of patterns that are common to two sequences, using the mathematical notion of irredundancy. This notion was studied first by Parida (1998) for the case of repeated patterns on a single sequence, and thereafter elsewhere (Apostolico and Parida, 2004; Pisanti et al., 2005; Apostolico and Tagliacollo, 2008). In Comin and Verzotto (2010), we extended the notion of irredundancy to the case of two sequences, to avoid the overcount of common patterns that “cover” the same region of a sequence. Indeed, one can easily show that there are lots of protein sequences that share an unusually large number of common patterns without conveying extra information about the input (see Table 4 below). To keep the article self-contained, here we summarize the basic facts already proved in Comin and Verzotto (2010).
Let s1 and s2 be two sequences, respectively, of m and n characters over an alphabet Σ. A character from Σ is called a solid character, while a don't care character ‘·’ equals and represents any character on Σ. Let s1[i, j] be the subsequence given by the j − i + 1 consecutive characters of s1 starting from position i. We call a suffix of s1 any subsequence of the type s1[i, m], with 1 ≤ i ≤ m.
A pattern p is a string over Σ(Σ∪{·})*Σ, thus having at least two solid characters: the first and the last character. A location [i, j] of s1 is an occurrence of p if s1[i, j] = p. A common pattern is a pattern that occurs in both s1 and s2.
In other words, property (1) of Definition 1 says that p2 has to extend p1 in composition (that is, p2 would be more specific than p1) and/or in length, and (2) indicates that p2 occurs in the same region of p1. We give an example of coverage in Figure 1. In this case, the occurrences of the common pattern ABA at [7, 9] on s1 and [1, 3] on s2 are covered, respectively, by the common patterns ABAA·C and A···ABA. Using Definition 1 we can now define an irredundant common pattern as a common pattern with an occurrence that cannot be deduced from the other common patterns without knowing the input sequences:
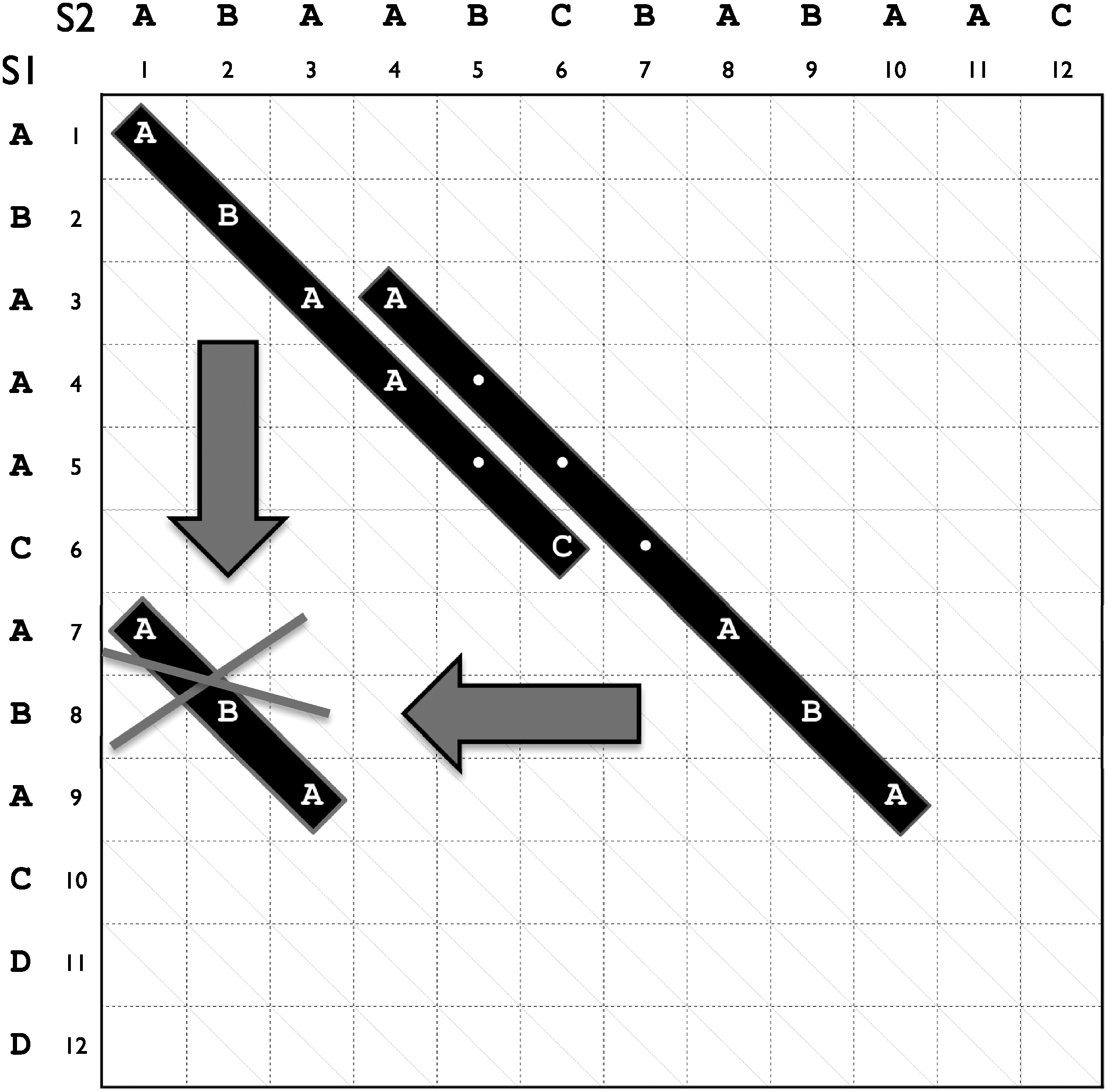
Coverage of pattern occurrences. Example of pattern occurrence coverage on the sequences s1 = ABAAACABACDD and s2 = ABAABCBABAAC of length 12. The occurrences of the meet ABA are covered, respectively, by the meets ABAA·C and A···ABA.
Clearly, a pattern that is not irredundant is called redundant.
As a result of Lemma 1 presented in Comin and Verzotto (2010), we have the following:
For example, in Figure 1, the common pattern ABA is the meet of s2 with the suffix s1[7, 12] of s1. However ABA turns out to be in any case a redundant pattern, therefore we need a more sophisticated algorithm to discover the whole class of irredundant common patterns, or Irredundant Class. In this regard, we refer the reader to the algorithm presented in Comin and Verzotto (2010).
As an immediate consequence of Theorem 1, we obtain that the number of irredundant common patterns of s1 and s2 is linear in the number of characters (or length) of the sequences:
Finally, we note that if s1 and s2 are identical, there is only one irredundant common pattern: the sequence itself. Moreover, we can extract the Irredundant Class in time O(z2 log z log |Σ|), where z = m + n, making use of the FFT in the step of searching for occurrences of the m + n − 3 meets described above.1
2.2. Scoring the irredundant class
Once the Irredundant Class
where Fp is defined as the number of occurrences of the pattern p in s1 and s2, and E[Fp] is the expected value of Fp. The value E[Fp] is computed combining the probability of each character a of p, extracted from the BLOSUM62 substitution matrix (Henikoff and Henikoff, 1992), with the length of the sequences:
where k is the length of p.
Unfortunately, the Irredundant Class (the name we will use for the general method in the rest of the article) seems to lack the positive-definiteness property, and therefore it must be treated as an indefinite kernel. In particular, following the work of Eichhorn (2007) for indefinite kernels applied to SVMs, we have that the Irredundant Class is in the case of weak non-positivity, and thus we need only to force the SVM optimizer to stop after a maximum number of iterations.
3. Why Resort to Irredundant Common Patterns?
The exhaustive detection of homologies in protein families and superfamilies leads to prohibitive computational methods, but on the other hand a low-complexity detection, for example using k-mers, would only consider a low-significant set of possible homologies, often overcounting the same information. These issues can be solved using an alternative method based on irredundant common patterns. Moreover, the automatic filter given by the notion of “non-redundancy,” or irredundancy, ensures us to select just those informative patterns that characterize the homologies of a pair of sequences.
We selected five algorithms of pairwise string similarity detection, used within the state-of-the-art protein classification methods, for the comparison with our method: Spectrum, Mismatch, Word Correlation (the core of Word Correlation Matrices), Local Alignment (namely the distance function given by all local alignments), and Smith-Waterman (the core of Pairwise).
3.1. Description of state-of-the-art pairwise string algorithms
In the following, we briefly explain the meaning of the selected algorithms on a pair of sequences s1 and s2, and then in the next subsection we estimate the redundancy, or information overcount, for each algorithm. Note that every algorithm computes a specific score for each extracted pattern, and then a global score is assigned to a pair of sequences using these pattern-specific scores.
In Spectrum (k), we count the number of occurrences for all the shared substrings (or consecutive subsequences) of length k on Σ in s1 and s2. In Mismatch (k, e), we count the number of occurrences for all the shared strings of length k on Σ in s1 and s2, and then we add each value to the other k-mers of which the meet has at most e don't cares. In Word Correlation (k), we compute a similarity score between all the k-mers of s1 versus all the k-mers of s2, and this is like to consider all the meets on Σ ∪ {·} of k-length substrings of s1 with k-length substrings of s2. In Local Alignment, we consider the global alignments between all pairs of substrings of s1 and s2 (given a scheme of scores for matches, substitutions, insertions, and deletions), that are all possible local alignments. Similar to Local Alignment, in Smith-Waterman, we take the best global alignment between all pairs of substrings of s1 and s2. In Irredundant Class, we consider all possible shared patterns on Σ ∪ {·} in s1 and s2, and then we avoid the contribution to be “overcounted” using the mathematical notion of irredundancy and selecting up to m + n − 3 patterns among the meets between all suffixes of s1 and s2.
3.2. Information overcount: from a theoretical perspective
For each method, we can now identify two characteristic phases: (1) pattern extraction and (2) pattern processing. We can think the output of phase (2) as a vector of pattern-specific scores, where each column represents just the score related to a single pattern.
For example, for Mismatch, (1) is the process of finding k-mers in the two sequences s1 and s2, while (2) is the multiplication of the respective number of occurrences of these k-mers in s1 and s2, where the number of occurrences of each k-mer is the number of times it appears with up to e errors. In this case, the output of phase (2) will be the vector of values resulting from the multiplications, and each column will represent a single k-mer. For Spectrum, (1) is the same as for Mismatch, but (2) is only the multiplication of the shared k-mer occurrences without any other preliminary process, and thus without error parameters. For Word Correlation, in (1) we individually find the k-mers of s1 and s2, and in (2) we compute a similarity score between all the possible pairs of these k-mers (one k-mer of s1 versus one of s2). For Local Alignment, (1) is represented by the extraction of all the substrings of the two sequences, while (2) is the global alignment of all the possible pairs of these substrings. For Smith-Waterman, (1) and (2) are the same as for Local Alignment, but in (2) we have also a max operation between all the computed values on the possible global alignments. For Irredundant Class, (1) is the extraction of all suffixes of s1 and s2, while (2) is the set of operations in which we compute the meets between a sequence and a suffix of the other one, and then we filter out the redundant ones.
Here we consider the information overcount as the number of outputs from phase (2) obtained taking into account the same pair of characters of s1 and s2 more than once:
Each output from phase (2), or component of the resulting vector, is intended as the score obtained comparing some pairs of single characters. For instance, after phase (2) of Spectrum we have a vector of values where each column represents the multiplication of the numbers of occurrences of a specific k-mer found in s1 and s2. These k-mers are overlapped in the two sequences by construction, and each component of the resulting vector represents at least k positions of each sequence. Therefore we use an information about the comparison of a shared position between s1 and s2 in more than one k-mer, and thus we store this information in more than one column of the final vector, resulting in an information overcount. We call the model that considers the information overcount as the Information Overcount Model.
Table 1 shows a comparison of the algorithms based on the Information Overcount Model, where we fixed a priori m ≥ n. The computation of these values is quite simple. For Irredundant Class, the meets between a sequence with all suffixes of the other sequence can be computed in a m × n grid, where each item represents the comparison of two different characters and each meet is a different diagonal of items from the top-left to the bottom-right part of the grid. Therefore, we have no information overcount. For Smith-Waterman, we have again no information overcount, because after phase (2) we consider only the best local alignment pattern that is comparing different characters in each position. For Spectrum, we could have at most n − k + 1 shared k-mers between s1 and s2. Thus, in this case, in s2 we have at most a coverage of k times (given by the k-mers) for each of the n − 2(k − 1) central positions, and at most a coverage of
Comparison of algorithms using the Information Overcount Model, with m ≥ n. Rows are listed from the best to the worst.
In Table 2, we present a comparison of pairwise computational complexity for the six algorithms described above, to give an idea of trade-off between information overcount (Table 1), computational complexity, and effectiveness in the classification of protein sequences (Table 3). These values were taken from the original papers.
Comparison of algorithms based on their pairwise computational complexity, where z = m + n. Rows are listed from the best to the worst.
Comparison of algorithms based on their mean ROC score over all experiments. Rows are listed from the best to the worst.
4. Experimental Results
We assess the effectiveness of the Irredundant Class method in the classification of protein families into superfamilies. This problem refers to the detection of sequence homologies in evolutionarily related proteins with low-sequence similarity, and is called remote homology detection.
Tests are based on the dataset described in Liao and Noble (2003),2 which uses the Structural Classification Of Proteins (SCOP)3 of Murzin et al. (1995), version 1.53. The data consist of 4,352 sequences grouped into 54 families and 23 superfamilies selected by Liao and Noble. For each family, proteins within the family are considered positive test examples, and proteins within the superfamily but outside the family are considered positive training examples; negative examples are chosen outside the fold, and were randomly split into training and test sets in the same ratio respect to the positive examples. Therefore this assessment consists of 54 experiments, each corresponding to a target family and having at least 10 positive training examples (taken from its respective superfamily) and at least five positive test examples (taken directly from the family), and no sequence homologies known a priori. In these experiments there is usually a much larger number of negative examples than of positive examples.
We compared the Irredundant Class with the state-of-the-art methods using as metric the receiver operating characteristic (ROC) score. For each experiment, given a ranking of the test example scores in output from the SVM, the ROC score is the normalized area under the curve that plots the number of true positive examples found as a function of the number of false positive examples detected (Gribskov and Robinson, 1996). This is like to plot the number of true and false positives found on a two-dimensional histogram (in abscissa the false positives, and in ordinate the true positives) for each different possible classification threshold based on SVM scores.
All methods compared are of discriminative nature, so we used a popular SVM software: the Gist SVM4 described in Noble and Pavlidis (2002), version 2.3. Experimental results of the other methods were taken from Saigo et al. (2004) and Lingner and Meinicke (2008).5
Table 3 shows the mean ROC scores, that is the average of ROC scores over all experiments, for the Irredundant Class and the other methods. These scores indicate that our method outperforms most methods in literature, and it is comparable to the state-of-the-art Local Alignment. For a more detailed view, the ROC scores distribution is illustrated for some methods in Figure 2. The Local Alignment (triangles) seems to perform slightly better than the Irredundant Class (squares), but the minimum ROC score of the Local Alignment is much smaller. In particular, the smallest ROC score of our method was obtained in experiment 15 of Liao and Noble (2003) with a value of 0.614, while all other methods got their lowest peaks in experiment 13 with very small values, for example 0.22 for the Local Alignment. To confirm this fact, Figure 3 reports the ROC scores distribution showing in detail the trend for all experiments, and evidencing that the Irredundant Class exhibits, in general, a more robust behavior than the other methods.
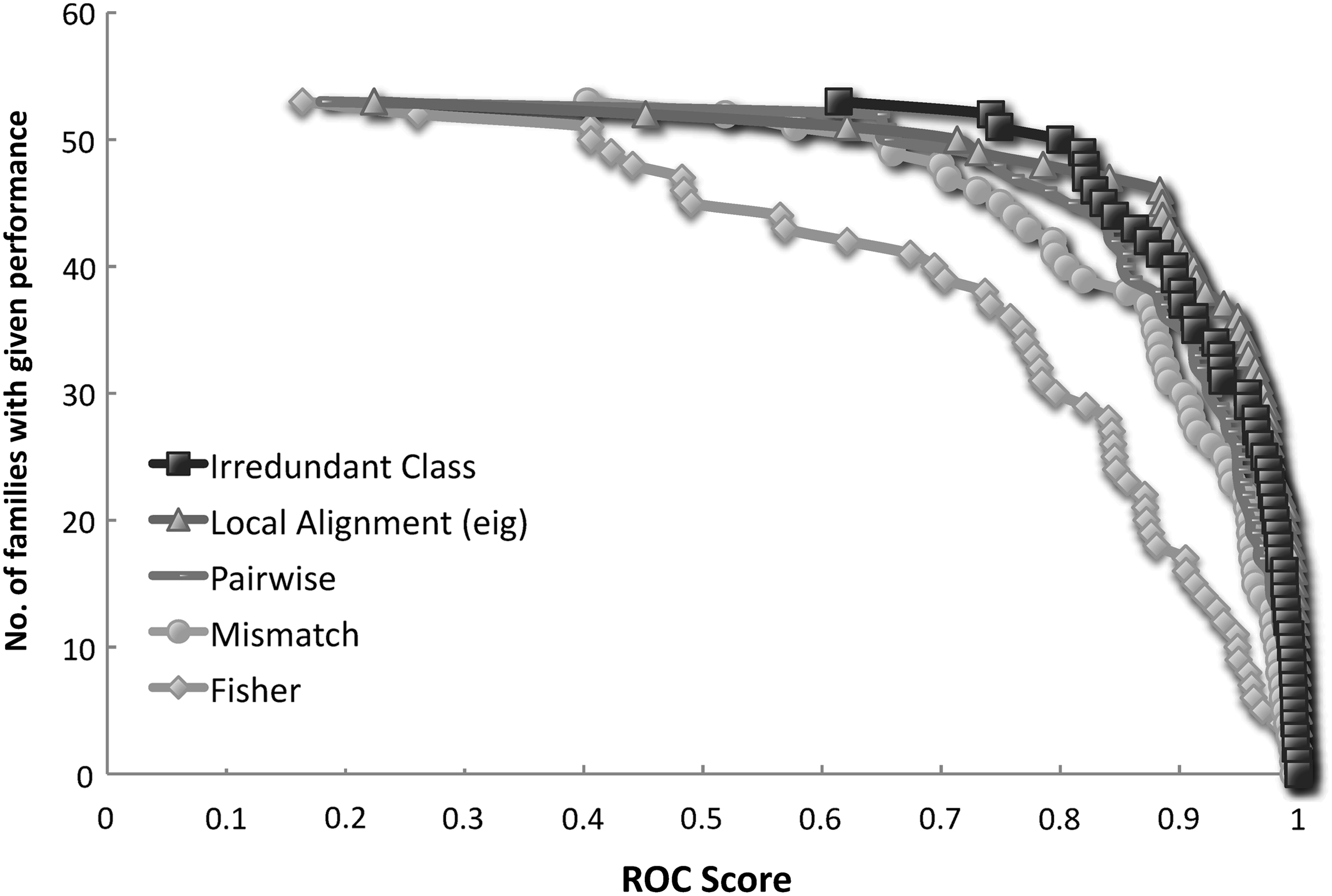
ROC scores distributions. ROC scores distribution for the Irredundant Class and the state-of-the-art methods.
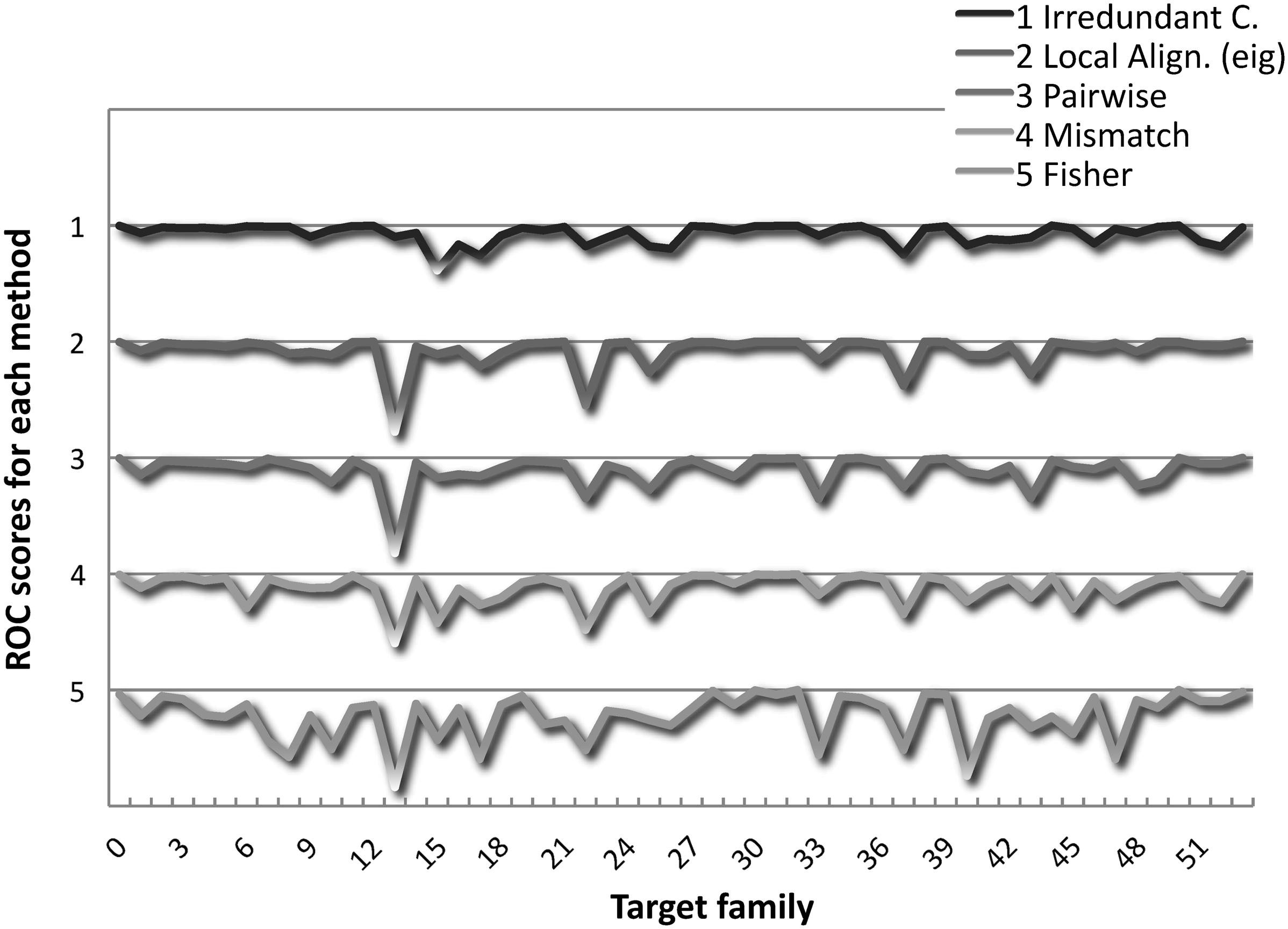
ROC scores family-by-family comparison. ROC scores across experiments.
Finally, Table 4 reports the number of irredundant common patterns against a less restrictive notion of patterns, the maximal common patterns introduced in Comin and Verzotto (2010), for 10 pairs of protein sequences taken from experiments. Results indicate that the number of irredundant common patterns
Number of irredundant
5. Conclusion
In this article, we study how the notion of irredundant common patterns can solve an issue that is rising in the field of string kernels, the remote homology detection. Almost all string kernels are based on patterns that are not independent, and therefore the associated scores are obtained using a set of redundant features. We specifically address this issue considering a particular class of patterns called irredundant common patterns. The method is based on the statistics of these patterns, and is called Irredundant Class. Results on benchmark data show that the Irredundant Class outperforms most of the string kernels previously proposed, and it achieves results as good as the current state-of-the-art method Local Alignment.
Despite its information properties, the Irredundant Class has a computational complexity that is much more than linear in the length of the sequences, and, in addiction, it processes the same characters of a single sequence a multiple number of times, due to pattern overlaps. Therefore, we plan to study a new notion of common patterns to manage these issues, and to better fit the problem of remote homology detection of protein sequences.
Footnotes
Acknowledgments
We thank Raffaele Giancarlo for having brought to our attention this problem and the related work. M.C. was partially supported by the Ateneo Project of the University of Padova and by the CARIPARO Project. D.V. was partially supported by the “Fondazione Ing. Aldo. Gini” during the preparation of this article, while visiting the University of California, Riverside.
Disclosure Statement
No competing financial interests exist.
1
This step, which is the most expensive in our procedure presented in Comin and Verzotto (2010), is described in detail in Fischer and Paterson (![]() ).
).