
Abstract

But here's what I want to tell you: He doesn't work in the lab. Of course that's not remarkable.
That was difficult for me to believe. You see, I've read his articles … masterful experiments! Who did them? I asked. He told me that he made a point of hiring someone who was a superb technician. Doesn't know a thing about biology, but that's OK. My friend tells him exactly what to do in the lab, and he does it.
It seemed rather strange to me, someone who knows nothing about experimental science directing someone who knows nothing about the subject of study. So I asked him, what inspired him to adopt this strange arrangement? He told me about his grandfather. His grandfather was an explorer. Discovered the South Pole. What inspired him was the fact that his grandfather was actually blind. He had a map in Braille and specially trained huskies. He'd point to the spot he wanted to go to, the dogs would sniff the map, and off they'd go!
Wait a second, wasn't he an
He had other talents too. He invented a remarkable machine for his wife, my friend's grandmother. She was a musicologist. His machine allowed her to feed in any musical manuscript, and it would tell her what composer's work it was most similar to.
“That's a remarkable machine!” I remarked. “But how could she tell if the machine was telling her the truth? “ “Well,” my friend answered. “The answer came out with E-values. The lower the E-value …”
“I know what E-values are, but that doesn't matter. Of course she could play the music herself to check up on the machine …”
“Well, no she couldn't do that.” My friend said. “She didn't know how to play any musical instrument. She was a musicologist, not a street musician! And anyway she'd been deaf since birth.”
OK, I think I might have lost you with that one. You don't believe that there could be a musicologist who relies totally on a machine to understand music. Perhaps you also don't believe that there could be a South Pole explorer who can see nothing of the world around him. I get your point, but actually, it's the first case that I find the most unbelievable: an experimental researcher who is blind to the means by which experimental information is obtained
Let's examine a situation that may seem more familiar.
Scenario: Nagging Doubts
Suppose that your experiments have focused your attention momentarily on a protein, Asr1156 (GenBank NP_485199), from the cyanobacterium Anabaena PCC 7120, that seems to be important in differentiation. What could this protein do? You go to your favorite web site and look up the annotation of the gene: “Hypothetical,” you're told. Oh, well. No help there. Back to your experiments.
Scenario (Part 2): Increased by A Provocative Blast
Suppose you go a step further, extracting the sequence of the predicted protein (perhaps this takes some irritating fiddling with the web site) and submitting it to Blast. Are there any similar proteins known? The result comes back: the protein is similar to the C-terminus of proteins from a large number of cyanobacteria. The protein is usually annotated as “an anti-sigma factor antagonist,” just the type of protein that might indeed be interesting with respect to differentiation! But you're concerned: Is the similarity incidental, perhaps just a conserved motif, or does it provide a sufficient basis to claim that your protein has the same activity as the rest? One web site says “hypothetical” and another suggests “anti-sigma factor antagonist” … how to decide?
Scenario (Part 3): Inflamed by A Remarkable Alignment
On most days, that difficult question would remain unanswered, but suppose that today you expend the considerable effort necessary to find the complete sequences of the similar proteins, download them, find a sequence alignment program, combine the sequences in a form acceptable to the program, and finally perform the alignment (Fig. 1A). It seems very peculiar that many proteins from organisms you know to be quite distant from each other have regions of very high similarity while other proteins appear truncated. For example, Anabaena variabilis (whose protein is just below Asr1156 in Fig. 1) and Prochlorococcus marinus MIT9303 (whose protein is sixth from the top) are as distant from each other as any two cyanobacteria can be, and yet their proteins have conserved residues not found in the truncated proteins of more closely related cyanobacteria. This strikes you as highly improbable and forces you to consider an alternative: Treachery! Perhaps the protein sequences are wrong! Perhaps what purports to be the start codons for the short open reading frames actually lie
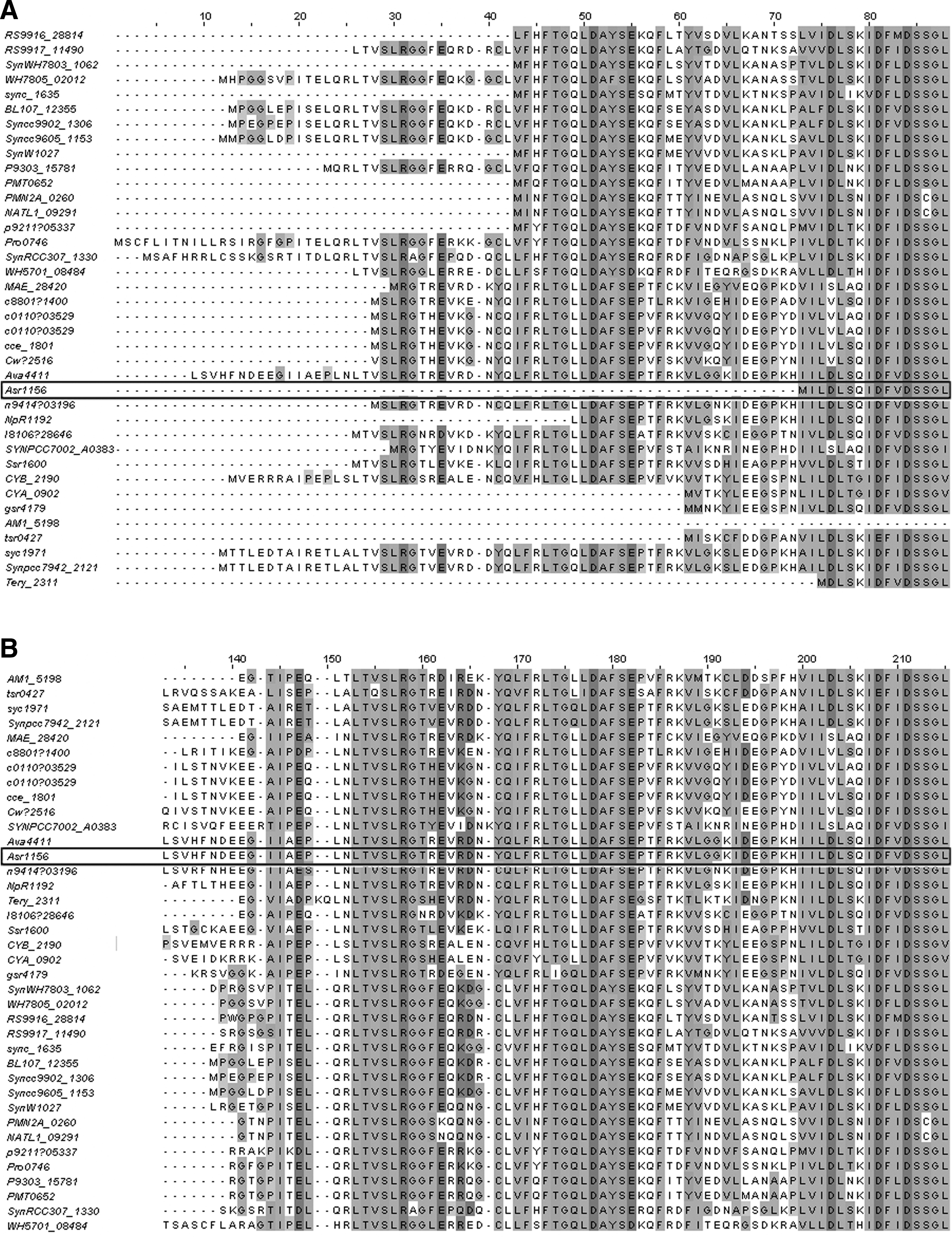
Alignment of sequences of cyanobacterial orthologs of Asr1156. The alignment was produced as shown later in Figure 6. To conserve space, only part of the alignment is shown. The line for the 70--amino acid protein p-Asr1156 is outlined.
Upon further reflection, you see an answer: you have the DNA sequences to go on, which are unlikely to be in error. Those sequences are the direct result of experiments, while the protein sequences are mere inferences. But the work required to get all those sequences and then to translate them…
Scenario (Part 4): Satisfied by An Astonishing Retranslation
Suppose that you are magically able to gather the upstream DNA sequences from the genes, virtually translate them in the genes' reading frames, and realign them, giving Figure 1B. Clearly, there is overwhelming sequence similarity going backwards from the nominal beginning of each protein up to a conserved isoleucine at position 95 in the alignment. Beyond that point, the sequences become dissimilar. The annotated start codons indeed must be wrong, and not only for Asr1156 but for every protein in the list! No other explanation can reasonably account for such sequence similarity in proteins whose organisms you know diverged well over a billion years ago. But why did the gene callers do such an abysmal job? You examine the DNA sequences: In most cases there is no conventional start codon between the conserved isoleucine and the next upstream stop codon. You realize that the beginnings of these genes are determined by something apart from one of the conventional start codons, most likely the codon of the isoleucine itself.
I have described a scenario where the end result is at odds with conventional wisdom, a result ordinarily hidden by the model of molecular biology built into our preexisting tools. In fact, in the case of one of the orthologs of Asr1156, there is strong experimental evidence that the protein begins with a conserved ATT (isoleucine) codon at the expected position (Sazuka and Ohara, 1996). Though not widely appreciated, ATT has been previously shown to function in some genes as a start codon (Binns and Masters, 2002).
All who work with their eyes open regularly encounter such situations, where things seem to be profoundly wrong. Most of the time, there is a trivial explanation, but sometimes pursuing such anomalies leads to new and powerful ways of looking at the world. The history of scientific research is strewn with unsought accidental discoveries (Roberts, 1989). It is important to note that those in computer science generally hold a different view of serendipity (Saunders and Thagard, 2005). In the natural sciences, where the goal is understanding the sometimes mysterious ways of nature, surprise can be the impetus for basic change, but in computer science, where the goal may be to find the best process to reach a given end, surprise is generally an unwelcome irritant. One should not expect that a collaborator charged primarily with building a tool should view an accidental discovery in the same way as one charged primarily with using the tool to understand nature.
Whether you recognize a serendipitous finding depends on the preparedness of your mind. Machines can't do it, nor can technical specialists unfamiliar with the burning issues in your field. But there's more to it than that. Following up the intriguing observation depends in part on the time required for the pursuit or even the ability to pursue. At many different points in the journey described above, most would have put aside their nagging doubts, filing away the curious circumstances or forgetting about them entirely, because a meaningful pursuit would require abilities that practically speaking most do not have. The sense that something is wrong is one of our greatest friends in research, but without the means to follow up an observation as it is made, with as low an energy barrier as possible, chance discoveries are lost. The precepts of our time remain unchallenged because they cannot be effectively challenged.
Choices In The Face Of Overwhelming Information
In the past several years, biology has been showered with riches—genome sequences, gene expression information, huge positional data sets—literally beyond imagining, since the common thread is that the information is too much to fit in any human head. To extract its value, one must use computational tools that in some ways illuminate but in other ways obscure the fundamental data. That's true of any tool, and the greatest insight comes from combining the abstractions a tool provides with an appreciation of the process through which the abstractions were obtained (Pevzner and Shamir, 2009). However, few biologists understand the computational tools they use (and the assumptions hidden within those tools), and fewer still can meaningfully grapple with the underlying sequences and numbers in a creative way that requires the most basic and flexible of computational tools—computer programming.
Biologists seem to be faced with a list of unpalatable choices:
One might imagine an additional choice, one that does not yet exist but should: that computer programming become as accessible to typical researchers as word processing, so that anyone can use it to manipulate bioinformation in creative ways with minimal activation energy.
Computation Versus Word Processing
The analogy of word processing is informative (Haigh, 2006). Just 35 years ago, word processing was confined to those who worked with computers professionally. Originally, commercial word processing programs were designed for pools of specialists within an automated office. This vision of the world did not last for long, as software companies discovered the more lucrative market of individual users. As a result, word processors evolved rapidly (Fig. 2) from clumsy line editors, through full screen editors driven by numerous key combinations, to complex programs that could interact productively with a great number of computationally naive users.
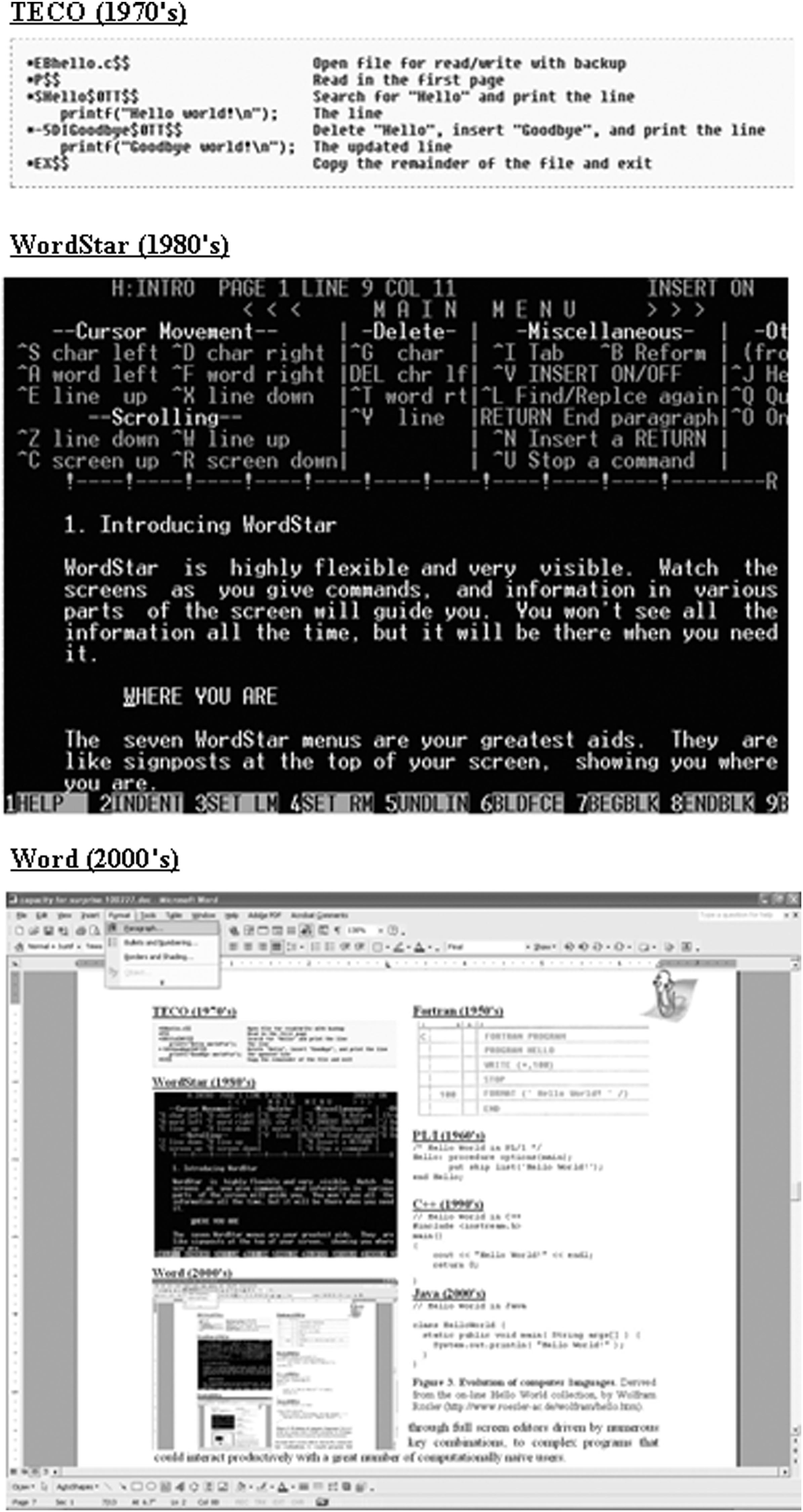
Evolution of word processors. Screen shots from three word processors/text editors.
Compare this progression of word processors with the succession of programming languages from the 1950s until today (Fig. 3). Computer languages have become far more powerful during that period, but they have not become significantly more usable by those new to the game. It is no easier today than it was 50 years ago for a novice to learn how to write simple computer programs. Probably less so.

Evolution of computer languages. Derived from the on-line Hello World collection, by Wolfram Rösler (http://www.roesler-ac.de/wolfram/hello.htm).
Is it truly more difficult to encompass the operations necessary to program a computer to do what we wish than it is to encompass the operations necessary to run a full-featured word-processor? Computer programmers will generally respond with a resounding "Yes!," but is that really so? If we wish to write complex applications that are efficient, robust, and resilient, then programming a computer is indeed complicated, intrinsically so. But if we wish to do those things that are simple for researchers to conceive of, then programming should not be so complicated. Just because typesetting a newspaper is inherently difficult, that's no reason why we should not have the capability to compose our thoughts on a word processor. We gain enormously living in a society where almost all can read street signs and write shopping lists, even though very few amongst us are Shakespeares.
In the end, however, I do confess that computer languages by their nature must be more complicated. After all, a word processor has only a limited number of things it can do (if we exclude the computer languages embedded in some), while, in contrast, connect a creative human, a digital computer, and a general purpose language, and what is there that they cannot do? How do we avoid being overwhelmed by the complexity that seems inevitable from a language that gives free rein to creativity? I'll return to this question in a moment.
Summary of the Problem
Fundamental changes in our views of the world come about through anomalous observations that challenge and ultimately break the prevailing models through which we filter perception (Kuhn, 1962). They are made possible by the ability to loosen the hold of the old paradigm and rearrange perceptions in a new way. The most basic discoveries are necessarily surprises, requiring a cohabitation in one mind of the surprising observation and the data (minimally filtered) that must be rearranged.
That's the way science is supposed to work. Today, the enormous amount of biological information—genomes, microarrays, data from other high-throughput experiments—has allowed research to make breakneck progress in increasing our understanding of how cells and organisms function. However, this rapid progress has come at a cost: a reduced connection between researcher and unprocessed data and the consequent reduced possibility for the most fundamental surprises. We gain in the short term. In the long run, we're in for trouble.
Most biologists are incapable of asking new questions directly about raw genomic information or any other mass data, because that would require going outside of fixed computational tools and making new tools. In short, it would require the ability to program a computer. We are stuck in our offices, cut off from the experiments that feed us the results we chew on, and unmindful of the surprising accidents that would rock our foundations. We are blind explorers, fixated on the South Pole but oblivious to new phenomena we don't know exist and don't know how to look for.
But how can biologists become computationally adept? It takes considerable effort to learn a programming language sufficiently well so as to build a new computational tool, and it takes a particular cast of mind to enjoy the process. Understandably, few biologists have made the investment. Human nature is unlikely to change. Biologists: It's not our fault!
How about machine nature? Machine interfaces
Towards A Solution: Human-Centered Programming
If the article stopped here, you might be excused for leaving discouraged. I hope, however, that you will instead take away the brighter note sounded at the end, that given sufficient incentive, powerful interfaces can be developed to engage humans with minimal computational experience. Our group has taken initial steps in this direction, developing an integrated knowledge and programming environment, called BioBIKE (
The main principles on which BioBIKE rests might be basic to any practical solution. First, knowledge is integrated with the means to manipulate it. Naive users should not be asked to assume that most difficult and most tedious of programming chores: finding data and converting it from one form to another. Second, utterances must be comprehensible at sight to those in a targeted community. An example is provided in Figure 4. Third, the language should cater to the needs of a specific clientele (in the case of BioBIKE, that would be those with questions in the domain of molecular biology). Questions or requests that are easy to pose in the language of the specified domain should be easy to express in the programming language. However, the language must still be capable of expressing anything within the realm of a general purpose language (perhaps with much more difficulty). Fourth, the language must be extensible by its practitioners, as a language can meet the needs of a community facing new phenomena only when that community is empowered to coin new tools and concepts. If a series of operations is found to be of general use, then a user should be able to package them into a chunk for use under a single name and that chunk incorporated into the language.

Example of a BioBIKE function. The function makes an alignment (shown in Fig. 1B) of the sequences of all orthologs of a protein, starting as many as 100 amino acids before the nominal beginning of each protein but going backwards only up to the first stop codon.
Unfortunately, these principles are not sufficient, even when assiduously followed, to make programming accessible in a practical sense to most biologists. A statement may make perfect sense when a user sees it but that same user may well be at a loss to construct it from scratch. We need the computer in Star Trek, one that can understand our intentions and figure out a way to satisfy them. Computer intelligence of that sort is far in the future. For now, the greatest progress may be made in developing a conversation between the language environment and the naïve user, where the interface does not need true intelligence but can nonetheless guide the user in the development of the needed tool.
Programming a computer will never be easy, because it is difficult to formulate questions that are both unambiguous and meaningful. However, formulating logical questions is what researchers do for a living. This is not the primary obstacle to enabling biologists to program the computer. That obstacle is the high activation energy that currently needs to be overcome to gain sufficient proficiency in a language to pose meaningful questions to a computer. So long as that obstacle remains in place, many biologists will continue to mush blindly forward, aided by easy-to-use programs built by many computer scientists.
I have tried in this article to address those two audiences. First, to those who study nature, if you are satisfied with your rate of progress, understand that traveling exclusively by highways offers speed but sacrifices most possible destinations. And if you despair at being able to depart from the smooth bioinformatics superhighway, trade that in for anger, and demand a language that enables you to analyze and manipulate mass biological information in a way that makes sense to you. Second, to those who are inventors of computational tools, consider the greater impact of devising a more general language that could potentially engage a vastly greater audience than those served by current computer languages, not one that aids a transition from a neophyte to a programmer like yourself, but a language that is an end point, one serving the needs of biological researchers and students. With this push and pull, a human-centered language might be developed sufficient to enable biologists to embrace computation as a basic tool, and we can thereby regain our historical connection with the raw data we study and regain the agility necessary to pursue surprising results to new biological insights.
Appendix
Computer programming as a tool in biology education
The disconnect from tangible reality that I argue now confronts researchers has long been a way of life for beginning students of molecular biology, much to their detriment. The advantages of learning by discovery rather than relying on lecture and textbooks are well documented (Wood, 2009), but molecular biology laboratories are expensive in both time and resources. Computational experiments offer an attractive alternative, and I have used BioBIKE to present university and high school students with a playground through which they can discover some concepts of molecular biology. At first, students must be led step by step through the process of asking useful questions and using BioBIKE to answer them, but it doesn't take long for them to gain increasing independence and the ability to strike out on their own.
I present here a highly abbreviated summary of an investigative tour that students without prior computational experience have used to discover concepts related to the nature of genes. You can find the complete tour by going to the BioBIKE portal (http://biobike.csbc.vcu.edu), clicking Guided Tours, and then What is a Gene?
Students bring with them all sorts of fanciful notions concerning genes that connect verbally with textbook descriptions but bear little resemblance with reality. It is important to help them build a mental picture of a real genome. In this tour, they use the genome of the cyanobacterium Anabaena variabilis (nicknamed Avar) as their laboratory. BioBIKE allows them to explore the genome (Fig. 5) and realize the problem confronted by the cell: how is it possible to discern the beginning and end of a gene from a continuous sequence of nucleotides?
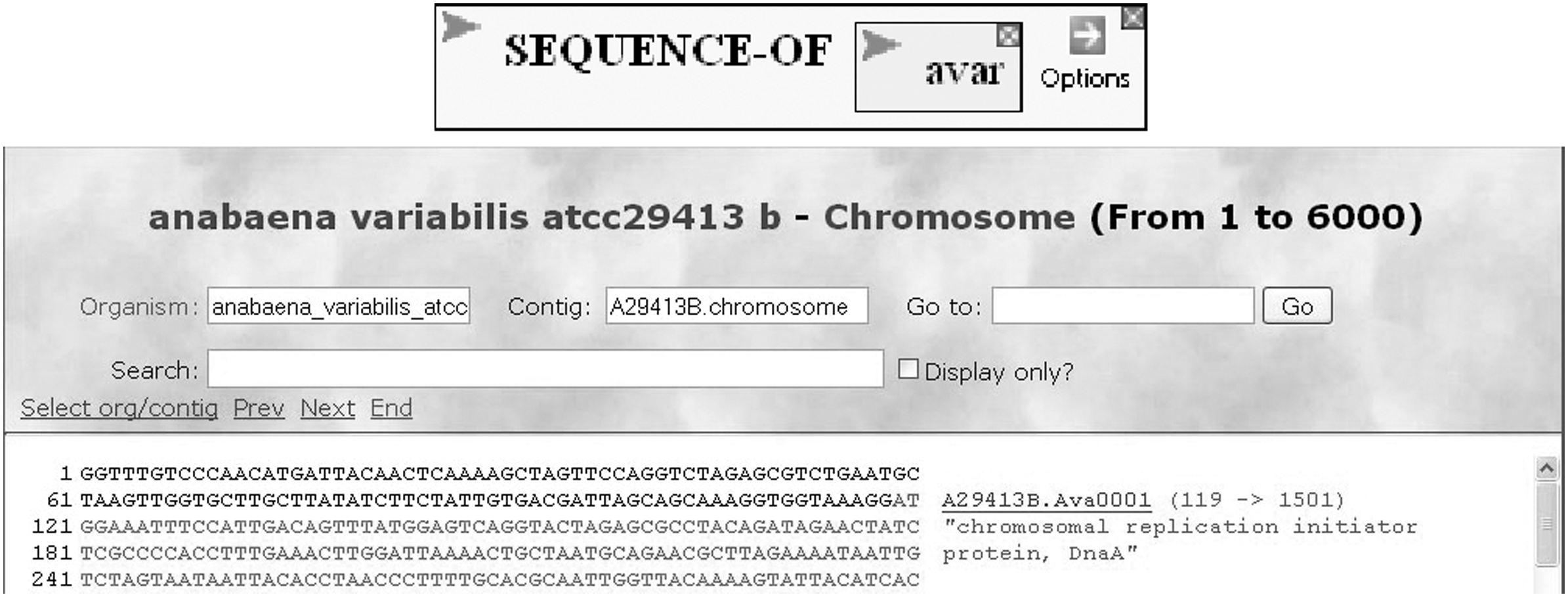
Display of a genome. The sequence of Anabaena variabilis (nicknamed Avar) is displayed using the indicated BioBIKE command. The student can explore the genome, displayed so that the putative termini of genes are superimposed on the sequence.
Drawing on the analogy of picking out sentences from English text, students consider two strategies: find internal signals in genes that mark their beginnings (analogous to capital letters), and find special sequences preceding genes (analogous to periods). To see if the internal signal hypothesis is true, the investigators experiment first with extracting the beginning of a single gene and then generalize the procedure over all genes of Avar. Seeing that there is a recurring pattern in the first three nucleotides, they focus on them, giving the set of opening triplets a name (Fig. 6).

Extraction of beginning triplets of all genes in a genome. A variable, genestarts, is defined as the set of triplets beginning all genes of Anabaena variabilis. Partial results of executing this definition are shown. Two unusual triplets are circled.
Many investigators come to the tour with an expectation of finding ATG at the beginning of genes, as is typically stated in textbooks. They are surprised to find that some genes begin with other similar triplets (Fig. 6). Do these triplets account for the beginnings of all genes? This can be tested by counting each one and comparing the sum to the number of all genes (Fig. 7). As it happens, 55 genes are missing, evidently beginning with some other triplet.

Comparison between the number of standard start triplets and the total number of genes. In the upper function, the number of start triplets defined as in Figure 6 that begin with any of the three triplets observed in the list are counted, yielding a list of three numbers. In the lower function, the total number of genes in Anabaena variabilis is determined.
What are they? Filtering the list of triplets beginning genes, retaining only those that do not match the pattern of “A”, “G”, or “T” followed by “TG” (Fig. 8), brings up a list of gene beginnings that are found to be associated with tRNA, rRNA, and other RNA-determining genes. In this way, investigators discover the concept of coding- and noncoding-genes. However, by counting the entire genome, they also find that there are many instances of “ATG” that do not begin genes. Some other feature must be important to signal the beginning of a gene.

Extraction of unusual start triplets. The set of start triplets defined as in Figure 6 are scanned for those that do not begin with A, G, or T followed by TG. Those that fit this criterion are returned as a list.
The second strategy they employ is to examine sequences upstream from genes, focusing on those genes that encode protein (Fig. 9A). At this stage, as in previous, investigators can go back to the genome sequence to make sure that the sequence extraction worked as expected. No obvious pattern emerges from inspection, so the upstream sequences are analyzed, counting the number of each nucleotide at each position, to obtain a Position-Specific Scoring Matrix (PSSM), as shown in Figure 9B. The complete count is done by the MAKE-PSSM-FROM function, and the results are plotted (Fig. 10A,C). It seems that there is a precipitous drop in A's and G's and corresponding rise in T's and C's before the beginning of genes. There's also a rise and fall in G's about 10 nucleotides before the beginning, and a strange up and down pattern in the A's.
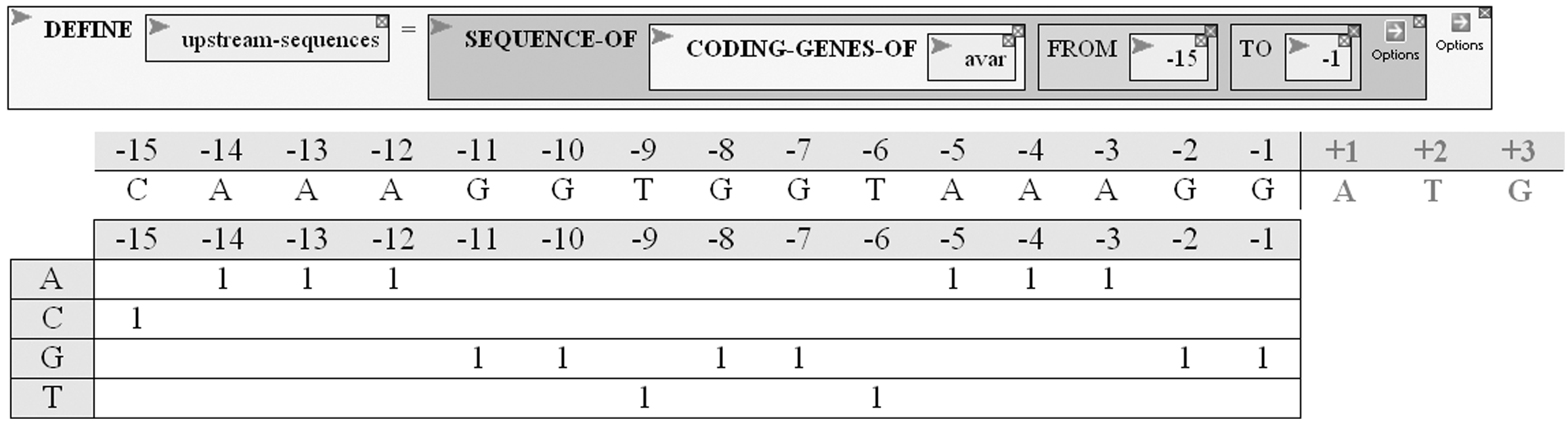
Analysis of regions upstream from coding genes. A variable is defined to contain the set of all sequences 15 nucleotides upstream from genes in Anabaena variabilis that encode proteins. Below the function is shown an intermediate stage in the formation of a position-specific scoring matrix of nucleotide frequencies of the upstream regions. The upper line shows the upstream sequence of the first gene considered, and the table below it shows how that region is tabulated. After all genes are tabulated, a frequency table will be generated from the counts.
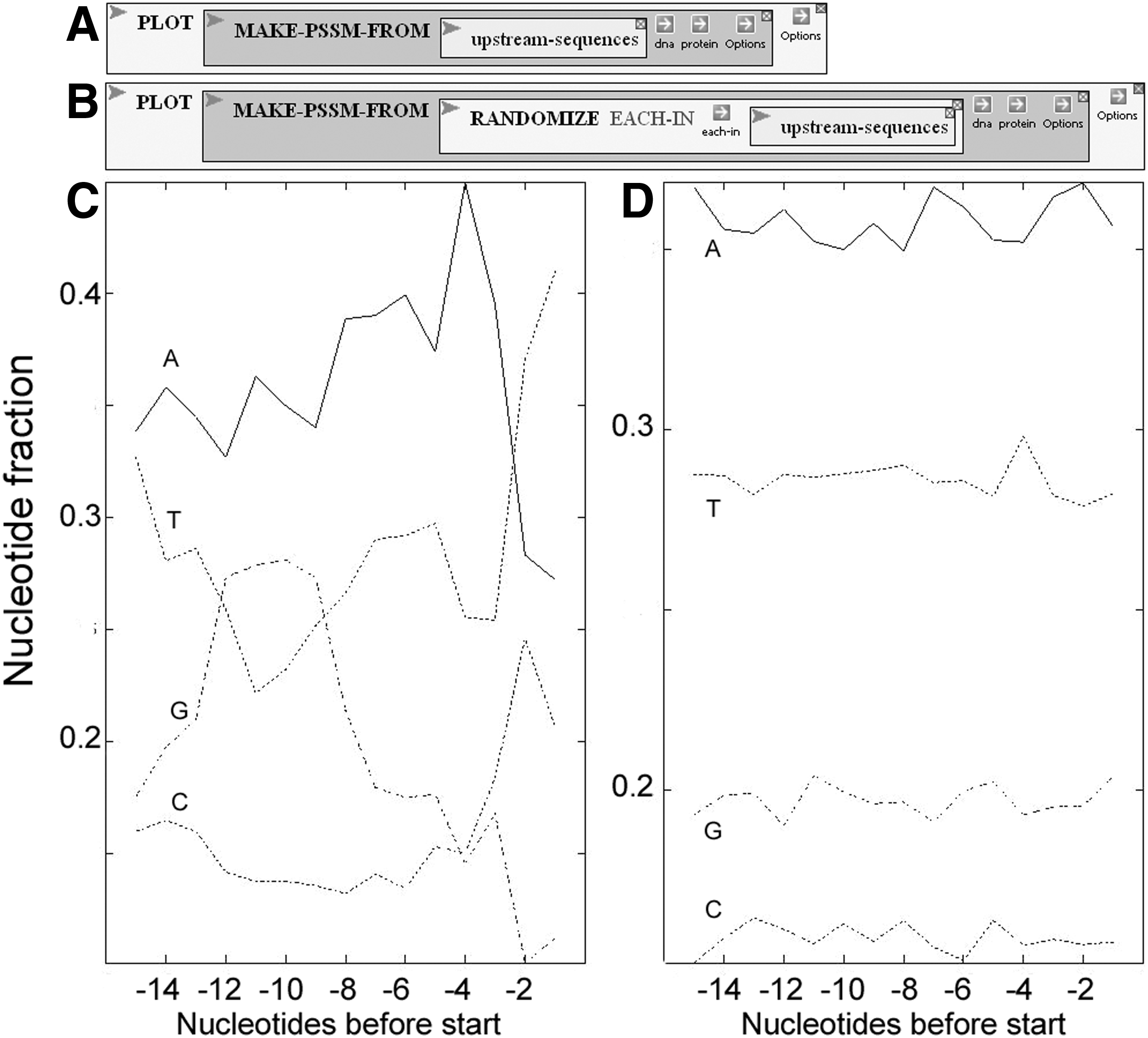
Visualization of upstream nucleotide frequencies.
How much of this is real and how much imagined, just the result of random fluctuation? Investigators can readily get a feel for what random fluctuation there is by repeating the plot a few times with randomized sequences (Fig. 10B), giving a typical plot shown in Figure 10D. Randomization is used in other investigative tours to give students an intuitive feel for what statistical tests do and how to interpret them.
The tour ends with some hints but otherwise a free-form invitation for investigators to attempt to try out the tools they have used to find interesting features near the
Needless to say, it is not of critical importance that students discover the diversity of start codons or the presence of ribosome binding sites typical of prokaryotic genes. What is important is that they discover—anything—so that they learn to make a habit of it. It is also not critical that they become comfortable with the latest bioinformatics tools (otherwise known as the obsolete tools of a decade from now). What is of lasting importance is a degree of comfort with uncertainty, a skepticism towards ideas and information, and an urge to check both in any way possible. Students learn that computation is a powerful ally in this regard and that they are capable of making use of it without losing control.
Integrating computation and quantitative thinking into the biology curriculum can go a long way towards bridging the current gap felt by many to lie between biology and numbers. A computer programming language that meets students more than halfway is necessary to encompass the great majority of biology students who avoid computation. Students who gain computational skills may emerge as a new generation of biological researchers, one that is able to encounter the surprises that are hidden in bioinformation and to pursue them to new insights.
Footnotes
Acknowledgments
This work rests on the efforts of J.P. Massar, Jeff Shrager, and Mike Travers, who first conceived of and built BioBIKE and provided philosophical outlets and responses to many of the thoughts expressed in this article. Thanks also go to John Myers, Peter Seibel, and Arnaud Taton, who provided much of the superstructure on which BioBIKE rests and to the many other participants in the project who sadly cannot be listed without going over the word limit. Thanks also go to anonymous reviewers for valuable comments. The work was supported by the National Science Foundation (grants DBI-0516378 and DBI-0850146).
Disclosure Statement
No competing financial interests exist.
*
In an informal survey of 6 departments of biology or microbiology in U.S. universities, only 4% of their members (out of 124) were reported able to write computer programs to the extent that they use them in their research.