
Abstract
Abstract
1. Introduction
However, experimentally determined protein complex data are still largely obtained through small-scale experimental techniques, which are time-consuming and tedious. On the other hand, numerous high-throughput methods (e.g., yeast-two-hybrid and protein chips) for detecting binary protein-protein interactions (PPI) en masse have recently become available. By making use of the high-throughput PPI detection methods, a graphical map of an entire organism's interactome can be constructed from the PPI data by considering the individual proteins as the nodes, and a physical interaction between a pair of proteins as a link between the two corresponding nodes. It has been observed that protein complexes generally correspond to dense subgraphs or cliques in the PPI networks (Spirin and Mirny, 2003; Tong et al., 2002), as proteins in the same complex tend to be highly interactive with one another. This has given rise to many computational methods to detect protein complexes from the pairwise PPI data—for example, MCODE (Bader and Hogue, 2003), MCL (Pereira-Leal et al., 2004), DPCLus (Altaf-Ul-Amin et al., 2006), DECAFF (Li et al., 2007b), COACH (Wu et al., 2009), and CMC (Liu et al., 2009), just to name a few. A comprehensive survey of such methods can be found in Li et al. (2010).
As mentioned, protein complexes are formed by simultaneous interactions between multiple proteins. The above methods are based on experimental data that describe pairwise interactions between two proteins. For detecting multi-protein interactions, an increasingly popular experimental method is Tandem Affinity Purification (TAP). TAP exploits immobilized “bait” protiens to capture “prey” proteins that that interact with them, with the preys themselves in turn serving as baits for other preys, thereby capturing multi-protein interactions. Recently, several computational methods have been developed to analyze TAP data for protien complexes. However, they typically convert the co-complex relationships in TAP data into binary interactions. The resulting pairwise PPI network is then mined for densely connected regions that are identified as putative protein complexes. In coverting the TAP data into PPI data, the bait-prey relationships and even the prey-prey relationships are regarded as candidate PPIs which can be scored by various reliability measurements (Collins et al., 2007; Gavin et al., 2006; Zhang et al., 2008). The candidate interactions with scores above a pre-determined threshold are the predicted as true PPIs.
Clearly, the value of this threshold directly impact the quality of the resulting PPI network. Most of the proposed methods determine this threshold based on reference protein-complex data. For example, in Zhang et al. (2008) and Pu et al. (2007), the reference protein-complex PPI map is constructed by treating the connections between all proteins within the same complexes as known positive interactions and the connections between proteins from different complexes as negative interactions.1 Obviously, it is not reasonable to assume that all protein pairs within a complex are positive interactions. In a recent study, Friedel et al. (2008) presented an unsupervised strategy for converting TAP data into PPI data. First, preliminary PPI networks are constructed by sampling the original TAP data. Preliminary complexes are then predicted from these preliminary networks using Markov clustering algorithm (Pereira-Leal et al., 2004). The final PPI network, called the bootstrap network in the paper, is then constructed linking two proteins to form a PPI if they occur together in at least one preliminary complex. Clearly, current methods for converting the multi-protein TAP data into binary PPI data for protein complex detection are fairly awkward. The additional step of converting the TAP data into PPI data not only introduces errors but also loses useful information about the underlying multi-protein co-complex relationships (Puig et al., 2001) that can be useful for protein complex identification (Geva and Sharan, 2011; Scholtens et al., 2005). Direct detection of protein complexes from the TAP data is therefore much desired.
Furthermore, protein complexes have been found to contain core-attachment structures in which the cores are the functional “hearts” of the protein complexes, and the attachment proteins assist the core proteins to perform subordinate functions (Dezso et al., 2003; Gavin et al., 2006). While there are also several recent studies detecting core-attachment structures from PPI networks (Leung et al., 2009; Srihari et al., 2010; Wu et al., 2009), we believe that the co-complex information in the TAP data could be exploited to better detect the core-attachment structures in the protein complexes.
In this article, we introduce a novel method called CACHET to detect protein complexes with
We performed extensive evaluation experiments on large-scale TAP data (Gavin et al., 2006; Krogan et al., 2006) and found that CACHET outperformed current methods in detecting protein complexes from these TAP data. In addition, we also evaluated the identified protein complexes and protein-complex cores using Gene Ontology (GO) functional information. The higher functional similarity found in the predicted complex cores is indicative of the biological significance of the protein complexes' structures detected by CACHET.
A newly proposed method called CODEC (Geva and Sharan, 2011) also detects protein complexes directly from TAP data as bipartite graphs. Our CACHET method works differently from CODEC in the following aspects. Firstly, CODEC detects dense bipartite subgraphs as protein complexes by iteratively adding proteins into seed subgraphs. Our CACHET selects protein-complex cores from bicliques and then simultaneously assembles all the attachments into cores to form the complexes. If we regard our cores as seed graphs, our approach for adding proteins into seed graphs to form protein complex is much more efficient than CODEC in terms of computation. Secondly, CACHET and CODEC address the issue of false positives in TAP data differently. CODEC approximates the reliability of bait-prey links mainly by the degree of the vertices. In contrast, CACHET exploits three state-of-the-art reliability measurements, such as Socio-Affinity (SA) (Gavin et al., 2006), Purification Enrichment (PE) (Collins et al., 2007), and Dice Coefficient (DC) (Zhang et al., 2008), which are demonstrated to assess the reliability of bait-prey relationships more accurately. As a result, our CACHET can detect protein complexes more efficiently, as well as with more biological significance (i.e., higher accuracy and homogeneity), than CODEC.
2. Preliminaries
As mentioned earlier, we represent TAP data as a bipartite graph G = (U, V, E), where U is the set of vertices for the baits, V the set of vertices for the preys captured by the baits in U, and E the bait-prey relationships. In addition, every edge
Next, let us introduce some relevant concepts for a bipartite graph G. From G = (U, V, E), a biclique G′ = (U′, V′, E′) is a bipartite graph such that for any two vertices
We define the reliability of a bipartite graph G = (U, V, E), R(G), as the average reliability scores of all the edges in E,
where w(u, v) is the edge weight (reliability score) of the edge (u, v), and δ(u, v) is 0 if the vertices u and v are identical and 1 otherwise. The latter is because a bait together with a specific antibody tends to tag the bait itself as a prey in pull-down experiments; we do not want to consider edges in the form of (v, v) for calculating the reliability score in Equation (1) (Geva and Sharan, 2011).
Similarly, we can also define a reliability score for each vertex l in G called ratioG(l) as follows:
We only use l's direct neighbors to compute its ratio score in Equation (2).
The Neighborhood affinity score (Bader and Hogue, 2003) is adopted to measure the similarity between two vertex sets. Given two bipartite graphs—G1 = (U1, V1, E1) and G2 = (U2, V2, E2)—we define their neighborhood affinity score, NA(G1, G2) as follows:
Figure 1 provides an example TAP bipartite graph. The baits v1 and v2 together with the preys v1, v4, and v5 form a biclique G′, with the reliability score
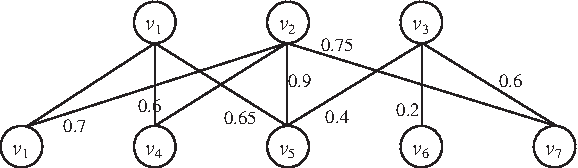
An example of a TAP bipartite graph.
3. Methods
Let us introduce our proposed algorithm CACHET to detect protein complexes and their core-attachment structures from TAP data. The key of our method is the detection of high-quality protein-complex cores. First, we compute the maximal bicliques in the TAP bipartite graph, since proteins within protein-complex cores are known to be highly interactive (Gavin et al., 2006). Next, we detect reliable protein-complex cores from the computed bicliques. Given the presence of false positive bait-prey interactions, we perform a filtering process to extract reliable bicliques (see Section 3.1). Since these reliable bicliques may have big overlaps with each other (i.e., they share many common proteins), we eliminate redundancy among these potential cores by computing a maximal independent set of them (see Section 3.2). The reliable and independent/non-redundant bicliques are predicted as the protein-complex cores. Finally, attachment proteins are included into the cores to form protein complexes with core-attachment substructures.
3.1. Mining reliable bicliques
We apply the FP-MBC algorithm (Li et al., 2007a) to compute the complete set of maximal bicliques from the bipartite TAP graph. We require that a detected maximal biclique should have at least Min_Bait baits and Min_Prey preys. In this study, both Min_Bait and Min_Prey are set as 2. We consider a biclique G′ to be reliable if its reliability score is greater than or equal to a threshold r, that is, R(G′) ≥ r. Reliable bicliques are likely to be protein-complex cores since most of their bait-prey interactions can be regarded to be true with respect to an appropriate threshold r.
Algorithm 1 shows the detailed steps for extracting reliable bicliques from the bipartite TAP graph GTAP = (UT, VT, ET). First, we compute the reliability scores for all the bait-prey edges in GTAP. We exploit multiple state-of-the-art reliability measurements such as Socio-Affinity (SA) (Gavin et al., 2006), Purification Enrichment (PE) (Collins et al., 2007), and Dice Coefficient (DC)(Zhang et al., 2008) for accurate edge-weighting. We preserve a biclique if it is already reliable (Lines 3–4). If a biclique is of low reliability, we iteratively delete a vertex with the minimum ratio score (Lines 7–8) to increase its reliability and obtain a reliable bilcique if possible (Lines 10–11). The vertex removal process is guaranteed to increase or at least maintain the overall reliability of the biclique in each iteration (for a theoretical proof, see the appendix in our supplementary material at http://www1.i2r.a=star.edu.sg/∼xlli/CACHET/CACHET.htm; the binary executables can also be found there). Supplementary Material is also available at www.liebertonline.com/cmb. During this process, we also require that the vertices to be removed are selected from an available node set to guarantee that all the detected reliable bicliques satisfy the size constraint (i.e., they should have at least Min_Bait baits and Min_Prey preys). For example, if a biclique G′ = (U′, V′, E′) has a bait set with the minimum size (i.e., |U′| = Min_Bait) and the prey set is still large enough (i.e., |V′| > Min_Prey), we should select a vertex from the prey set and then remove it. The available node set in G′, denoted as A(G′), is defined in Equation (4):
Note that our iterative removal process (Lines 6–9) will stop with the biclique in question being either preserved or discarded. If a biclique is reliable after the removal of some vertices, it will be preserved in Line 11 and the iteration stops. Otherwise, it will be discarded if it has reached the minimum size (i.e., A(G′) = ϕ), and yet it is still not reliable.
Mining reliable bicliques
3.2. Mining maximal independent bicliques
The detected reliable bicliques may overlap with each other. As such, we need to select a maximal subset of independent/non-redundant bicliques as our protein-complex cores. To do so, we construct a redundancy graph, denoted as RG = (VRG, ERG, W), where the super vertex set
The vertex weighting function W computes the significance of reliable bicliques as follows. First, we count the frequency of each vertex/protein in all the reliable bicliques to indicate their respective importance, given that proteins that appear in multiple modules are likely to be more biologically important (Navlakha and Kingsford, 2010) than those that do not. Equation (5) below computes w(u) as the importance score of the vertex/protein u, where the frequency of u, freq(u), is the number of reliable bicliques involving u:
We refine the reliability score for bicliques in Equation (1) by considering the importance of vertices to evaluate the significance of reliable bicliques:
where G′ = (U′, V′, E′) is a reliable biclique, the edge weight w(u, v) is the edge reliability (Collins et al., 2007; Gavin et al., 2006; Zhang et al., 2008), and the vertex weight w(u) is the vertex u's importance as computed in Equation (5).
Given a redundancy graph RG = (VRG, ERG, W), our aim is to mine for an independent set (IS) in RG in which no two reliable bicliques are similar. This is a maximum independent set (MIS) problem. To conserve the more significant bicliques, we maximize the weight sum of the super vertices in the resulting independent set. Our MIS problem can thus be modeled as a maximum weighted independent set (MWIS) problem, as defined in Equation (7):
Here, IS is an independent set of RG and the resulting set SC—the maximum weighted independent set—contains reliable and independent bicliques which form the set of our predicted protein-complex cores. Given a super vertex
Maximum weighted independent set
The first heuristic (H1 in Line 4) (Sakai et al., 2003) greedily selects the super vertex with the maximum
3.3. Detecting protein-complex attachments
Given that we have detected independent reliable bicliques as protein-complex cores, let us now identify the attachment proteins. For a protein-complex core Gc = (Uc, Vc, Ec) detected from the bipartite TAP graph GTAP = (UT, VT, ET), we detect its bait-attachments and prey-attachments as follows. Given a prey protein
The resulting protein complexes are then formed by combining the protein-complex cores and their bait-attachments and prey-attachments (Supplementary Material Figure 1 depicts how our CACHET works on an example TAP graph).
4. Experimental Results
Before we present our experimental results, let us introduce the evaluation metrics and the datasets used in our experiments.
4.1. Evaluation metrics and experimental data
Given a predicted complex p = (Vp, Ep) (Vp comprises the proteins in the core and attachments of p) and a real complex b = (Vb, Eb), we use their neighborhood affinity score, NA(p, b), to determine how well they match with each other. We consider them to be matching, if NA(p, b) ≥ ω. While ω has been typically set as 0.2 in numerous previous studies (Altaf-Ul-Amin et al., 2006; Bader and Hogue, 2003; Li et al., 2007b; Wu et al., 2009), some may feel that the value of 0.2 is too low and liberal. Therefore, we also tried different values of ω from 0.2 to 0.5 for a more comprehensive evaluation. Please note that 0.5 is quite a high and stringent value. For example, given two complexes both with six proteins, they are only considered to be matched with each other if they share at least five common proteins.
For overall comparison of the methods, we use Precision, Recall, and F-measure to evaluate the performance of various methods (Chua et al., 2008; Wu et al., 2009). Let P and B denote the sets of complexes predicted by a computational method and those in the benchmark complex set, respectively. Let Ncp be the number of predicted complexes in P that match at least one benchmark complex, and Ncb be the number of benchmark complexes in B that match at least one predicted complex. Equations (8) and (9) give the definitions of Precision, Recall, and F-measure:
Note that the matching pairs of complexes may have different NA scores that indicate different degrees of matching. This issue is not taken into consideration in the above definitions. As such, we have also conducted a protein-level evaluation (Liu et al., 2009) for the various methods (for details, see Table 3 in Supplementary Material).
We also use GO to evaluate the functional homogeneity of predicted protein complexes based on p-values (Li et al., 2010). The p-value of a predicted complex C with respect to a functional group F is defined in equation (10):
where C contains k proteins in F, and |V| is the total number of proteins in the TAP data. The p-value of a predicted complex is the smallest p-value over all the possible functional groups. As such, a predicted complex with a low p-value indicates that it is enriched by proteins from the same function group and it is thus likely to be a true protein complex. In our experiments, the p-values (with Bonferroni correction) of identified complexes are calculated using SGD's GO::TermFinder (Boyle et al., 2004).
We applied CACHET on two public large-scale TAP data, namely, the data of Gavin et al. (2006) and the data of Krogan et al. (2006). Let us denote Gavin et al.'s data and Krogan et al.'s data as Ggavin = (Ug, Vg, Eg) and Gkrogan = (Uk, Vk, Ek), respectively. Ggavin contains 1993 baits, 2670 preys, and 19277 bait-prey links, whereas Gkrogan contains 1999 baits, 5202 preys, and 47204 bait-prey links. We also generated a dataset by combining Gavin et al.'s data with Krogan et al.'s data, denoted as Gcombined = (Uc, Vc, Ec), where Uc = Ug ∪ Uk, Vc = Vg ∪ Vk and Ec = Eg ∪ Ek. The combined dataset contains 2850 baits, 5337 preys, and 63631 bait-prey links. Table 1 shows the relevant statistics about these three datasets. The benchmark protein complexes were downloaded from Wodaklab (Pu et al., 2009). The catalogue is called CYC2008, and it contains 408 manually curated protein complexes. (GO was downloaded from http://geneontology.org.)
4.2. Evaluation of predicted protein complexes
We focus on presenting our results on Gavin et al.'s data in this subsection. Similar results were obtained on Krogan et al.'s data, and they can be found in Section 3 in Supplementary Material. We will show the results of CACHET on the combined data in Section 4.6.
First, we computed the results using the three different MWIS heuristics with the parameter r set as 0.7 (see Algorithm 1 for r; we have also performed the sensitivity study of r in Section 4.4). The second column of Table 2 shows the number of protein complexes predicted by different MWIS heuristics. H1 and H2 predicted nearly the same number of protein complexes and achieved similar Precision. Compared with H1 and H2, H3 obtained fewer protein complexes, but achieved a higher Precision with similar Recall and higher F-measure. Given that time complexity is a practical constraint that we have considered, we only report the results using H3 in our CACHET hereafter, as CACHET with H3 detects protein complexes most efficiently.
We compared our CACHET with the existing state-of-the-art methods, namely, MCODE (Bader and Hogue, 2003), MCL (Friedel et al., 2008; Krogan et al., 2006; Pereira-Leal et al., 2004), DC-CM (Zhang et al., 2008), COACH (Wu et al., 2009), and CODEC (Geva and Sharan, 2011) (CODEC has two different weighting schemes, denoted as CODECw0 and CODECw1. For details, in terms of F-measure and p-values, see Geva and Sharan (2011).
As we need to transform the TAP data to PPI networks to run the traditional algorithms (MCODE, MCL, and COACH) in these networks, the method that we have used to convert TAP data to PPI networks (Zhang et al., 2008) can be found in Section 2 of Supplementary Material. Note that applying different reliability schemes will result in different PPI networks during the conversion process. That is, these three methods (MCODE, MCL, and COACH) may obtain different results using different reliability schemes (see Table 2 in Supplementary Material, which shows the PPI networks converted from TAP data and indicates that applying different reliability schemes on the same data will result in different PPI networks).
Figure 2 shows the F-measure of various methods using three different reliability schemes on the data of Gavin et al. Our CACHET with SA scores achieved the highest overall F-measure. CACHET also achieved the highest F-measure when using SA and DC scores when compared to the other methods using the same scoring schemes. When using PE scores, CACHET did not perform as well as MCL, which has a F-measure of 48.6% and is the best performer of existing methods over all scoring schemes. Our CACHET with SA scores is still 4.8% higher than MCL with PE scores.

Comparison among various methods employed on Gavin et al.'s data using different reliability schemes.
As SA scores are most widely used, Table 3 shows the details of the comparison among various methods using SA scores (some methods are independent of reliability measurements, such as DC-CM, CODEC, and the original results “Gavin” and “Gavin-Core” from Gavin et al. [2006]). The F-measure of our CACHET is 53.4%, which is 7.9%, 8.3%, and 11.4% higher than that of MCL, COACH, and CODEC w1, respectively, the top three performers of the existing methods. Furthermore, CACHET is shown to perform better than CODEC in terms of all three measures, although CODEC also models TAP data as a bipartite graph as we do. In addition, we also show the running time of our CACHET on Gavin et al.'s data in Section 6 of Supplementary Material, which indicates that CACHET is computationally more efficient than CODEC.
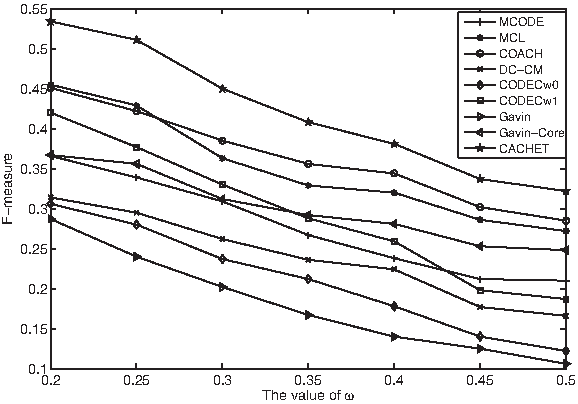
Finally, Figure 3 shows the F-measures of various methods when ω is varied from 0.2 to 0.5. CACHET consistently outperformed existing methods. This implies that the superiority of our CACHET is independent of the values of ω.
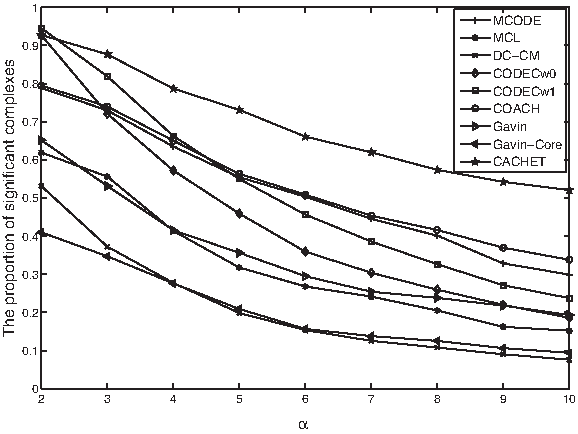
The proportion of significant complexes predicted by various methods.
We also investigate the proportion of protein complexes detected by the various methods that have statistically significant functional homogeneity. A protein complex has statistically significant functional homogeneity if its p-value is less than 10−α the larger the α, the more likely that the complexes are genuine. Figure 4 shows the proportion of significant complexes under different values of α; In this figure,2 CACHET and CODEC (with both weighting schemes) are shown to achieve the highest proportion of significant complexes when α = 2. As the value of α increases, the superiority of our CACHET becomes obvious. For example, CACHET has 77.0% significant complexes, 10.7% higher than the second best (i.e., CODECw1 with 66.3%) when α was set as 4. Furthermore, CACHET has 46.8% significant complexes, 13.0% higher than the second best (i.e., COACH with 33.8%) when α was set as 10. This shows that complexes predicted by CACHET are enriched by proteins with the same functions and are thus more likely to be true complexes.
F-measure of various methods when ω is varied from 0.2 to 0.5.
4.3. Evaluation of predicted protein-complex cores
Proteins within the complex cores have been found to have higher functional similarities and are more co-localized than the attachment proteins (Dezso et al., 2003; Gavin et al., 2006). For evaluation, we can calculate a functional similarity score for a core (or a protein complex) by averaging the semantic similarities among all bait-prey pairs (Wang et al., 2007). Table 4 shows the average functional similarity for all cores and protein complexes.
The functional similarities in Table 4 were calculated using two subontologies of GO (biological process and cellular component). The protein-complex cores generated by all three MWIS heuristics were found to have higher average functional similarities than the corresponding protein complexes. This demonstrates that our detected cores are more functionally consistent and likely to be more biologically significant. Figure 5 shows two examples to illustrate the organization of our predicted complexes. In Figure 5A, our identified core was reported in Gavin et al. (2006), and the predicted complex also matched well with the histone deacetylase complex (all eight proteins in our identified complexes are involved in the benchmark histone deacetylase complex). The protein YPL139C was identified as both bait-attachment and prey-attachment, and it is required to bind the protein YNL330C in the core for its activity. The protein YNL097C, which is predicted as another bait-attachment, has also been shown to be a possible component of the histone deacetylase complex (Gavin et al., 2006, 2002). In Figure 5B, the identified complex contained 14 proteins, which covered 10 of the 12 proteins in the benchmark DNA-directed RNA polymerase II complex (Cramer et al., 2000). The other four proteins in gray (YDL115C, YGR005C, YGR186C, and YML010W) are novel to our benchmark complex. However, YDL115C has already been demonstrated to belong to the DNA-directed RNA polymerase II complex (Krogan et al., 2006). The other three proteins identified as attachments were all annotated with the term GO:0006366 (transcription from RNA polymerase II promoter), which is also closely related to the function of this complex. This example indicates that our CACHET may be used to detect the potential knowledge absent in current reference data. Our identified core with the seven proteins was also validated in Gavin et al. (2006).
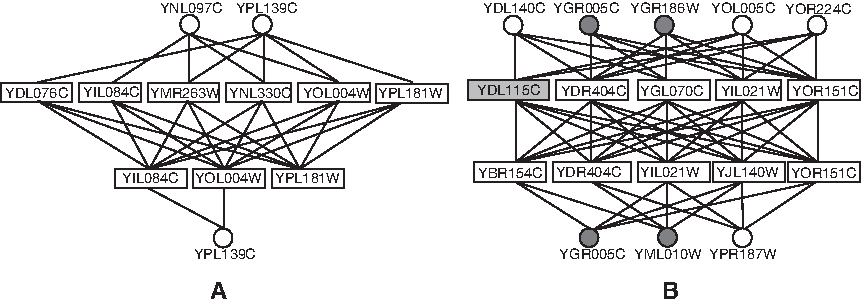
The organization of our identified protein complexes. The predicted complexes are histone deacetylase complex in (
4.4. Effect of the parameter r
The reliability parameter r in Algorithm 1 determines the number and quality of the protein-complex cores detected by our CACHET. We investigate how the variation of r affects the performance of our CACHET. Figure 6 shows the Precision, Recall, and F-measure of CACHET on Gavin et al.'s data under different values of r.
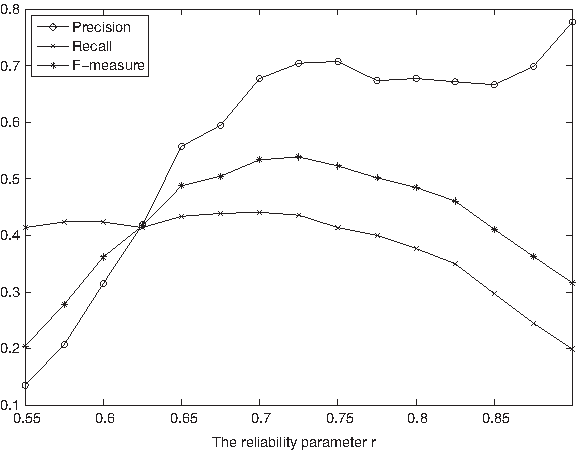
Performance of our CACHET method used on Gavin et al.'s data under different values of r.
Fewer false positive bait-prey links are involved in our identified complexes as we increase the reliability value of r, as expected. The predicted complexes are thus more likely to be true complexes, as reflected by the ascending trend of the precision of CACHET when r becomes larger, as shown in Figure 6. On the other hand, when r becomes too large, CACHET may filter away some true positive bait-prey interactions. As such, it will predict a small number of complexes with a low recall but achieving a very high precision. Overall, the resulting curve for the F-measure in Figure 6 has three distinct ranges. It increases significantly in the first range (
4.5. Impact of reliability indexing and redundancy filtering
Recall that indexing the reliability of bait-prey links to identify reliable bicliques (denoted as Step I; see Section 3.1) and filtering redundancy amongst the detected reliable bicliques (denoted as Step II; see Section 3.2) are two key steps in CACHET. To see the impact of reliability indexing and redundancy filtering, we performed experiments to run CACHET without each step, respectively. In Table 5, CACHETwoI and CACHETwoII denote CACHET without Step I and Step II, respectively. CACHETwoI was observed to achieve a much lower precision than CACHET and CACHETwoII, indicating that detecting reliable bicliques does filter false positives effectively and increase the reliability of resulting protein complexes. Although CACHETwoII has the highest recall, it predicts protein complexes with huge redundancy (e.g., 4576/188 = 24.3 predicted complexes match a real one on average). The results showed that these two steps in CACHET are indeed essential to filter potential false positive interactions and reduce redundancy among protein-complexes.
4.6. Running CACHET on the combined data
Currently, Gavin et al. (2006) and Krogan et al. (2006) have already provided comprehensive TAP data. However, they just produced quite different descriptions of the yeast interactome due to the fact that they exploited different purification techniques and utilized a different number of baits in their purifications. Therefore, combining these two data sources will definitely improve the coverage of the data for better identifying protein complexes (Friedel et al., 2008; Hart et al., 2007; Pu et al., 2007).
Assuming that Ggavin = (Ug, Vg, Eg) and Gkrogan = (Uk, Vk, Ek) denote Gavin et al.'s and Krogan et al.'s data, respectively, Gcombined = (Uc, Vc, Ec) is the resulting combined data, where Uc = Ug ∪ Uk, V
c
= Vg ∪ Vk, and Ec = Eg ∪ Ek. To index the reliability scores of edges in Ec, it is a possible solution to re-compute these scores based on the purification records in the combined data. Here, we adopt the strategy used in Collins et al. (2007) to assign reliability scores for bait-prey links in the combined data based on their original scores in individual datasets. For an edge
We compared our CACHET with existing methods: MCODE, MCL, COACH, the bootstrapping method (BT) (Friedel et al., 2008), Pu et al.'s method (Pu et al., 2007), and Hart et al.'s method (Hart et al., 2007). Since the last three methods are independent of scoring schemes, we first show the F-measure of CACHET, MCODE, MCL, and COACH using various scoring schemes in Figure 7. The result is very similar to that of Gavin et al.'s data (Fig. 2). In terms of using individual scoring schemes, CACHET achieved the highest F-measure when using SA and DC scores, and performed equally well as MCL when using PE scores. In particular, CACHET with SA scores achieved the highest F-measure as compared to other methods with any scoring schemes. For example, CACHET with SA scores has an F-measure of 61.5%, (5.5% higher than MCL with PE scores), which is the best performer by combining existing methods and scoring schemes.

Comparison among various methods employed on the combined data using different reliability schemes.
CACHET with SA scores predicted 449 protein complexes from the combined data (the reliability threshold is set as 0.7, which is the same as on the Gavin et al.'s data). Compared with the results from individual data, CACHET achieved higher Precision, Recall, and F-measure on the combined data as shown in Table 6, indicating that CACHET can effectively exploit the biological knowledge from multiple data. Table 7 shows the comparison details between our CACHET using SA scores and other methods on the combined data. While MCL, BT, Pu, and Hart were shown to obtain relatively high recall, our CACHET gained much higher Precision, and thus obtained the highest overall F-measure. Our higher Precision indicates that the complexes predicted by CACHET are more accurate, implying that our unmatched complexes are more likely to be novel true complexes. We also show the F-measure of the above methods with different values of ω in Figure 8 and observe that our CACHET consistently achieved the best overall performance even with more stringent matching criteria.

Performance of our CACHET method used on Gavin et al.'s data under different values of ω.
Finally, for functional homogeneity, Figure 9 shows the proportion of statistically significant complexes from the combined data under different values of α. Our CACHET is again observed to consistently achieve the highest proportion of statistically significant complexes. For example, when α was set as 2, CACHET produced 89.8% significant complexes, which is 6.4%, 18%, and 23.3% higher than COACH, Hart, and Pu (the top three performers), respectively.
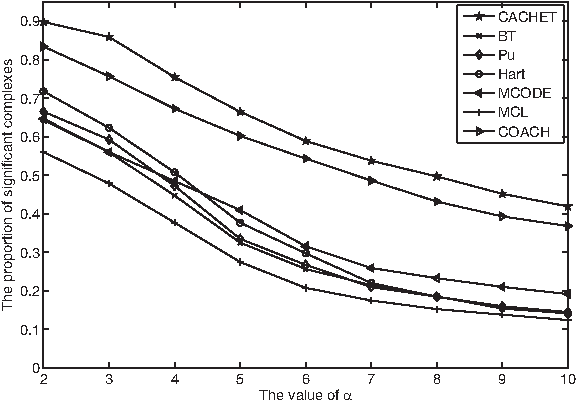
The proportion of significant complexes predicted by various methods from the combined data.
5. Conclusion
In this article, we have proposed a novel algorithm CACHET to detect protein complexes as well as their core-attachment structures from TAP data. CACHET first computes all the maximal bicliques from the TAP bipartite graph. It detects high-quality protein-complex cores from the computed maximal bicliques by extracting reliable bicliques and removing redundancy among them. Then, CACHET forms protein complexes by incorporating attachments into cores.
CACHET significantly outperformed existing methods in terms of the F-measure and functional homogeneity of predicted complexes. Experimental evaluation on our identified protein complexes and cores also demonstrated the following advantages of our CACHET. First, as CACHET detects protein complexes directly from TAP data, it is able to exploit the co-complex information in the TAP data and avoid the step of converting the TAP data into a binary PPI network. Second, the protein complexes predicted by CACHET are with core-attachment structures, which provide useful information for revealing the inherent organization of the detected protein complexes.
Footnotes
Acknowledgment
This article was supported by Singapore MOE AcRF grant no. MOE2008-T2-1-074.
Disclosure Statement
No competing financial interests exist.
1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.