
Abstract
Abstract
In comparative genomics, differences or similarities of gene orders are determined to predict functional relations of genes or phylogenetic relations of genomes. For this purpose, various combinatorial models can be used to specify gene clusters—groups of genes that are co-located in a set of genomes. Several approaches have been proposed to reconstruct putative ancestral gene clusters based on the gene order of contemporary species. One prevalent and natural reconstruction criterion is consistency: For a set of reconstructed gene clusters, there should exist a gene order that comprises all given clusters. For permutation-based gene cluster models, efficient methods exist to verify this condition. In this article, we discuss the consistency problem for different gene cluster models on sequences with restricted gene multiplicities. Our results range from linear-time algorithms for the simple model of adjacencies to NP-completeness proofs for more complex models like common intervals.
1. Introduction
One approach for the reconstruction of phylogenetic scenarios or for the comparison of genomes is the examination of the genetic material on the level of the DNA, RNA or protein sequences. Another possibility is to study the genomic structure. On this higher level, one commonly considers the order of the genes or other markers within the genome. Genes belonging to the same gene family are represented by the same identifier. To simplify matters, the term “gene” will be used to refer to the corresponding gene family identifier. One simple way to model genomes is to use permutations. However, this approach includes the assumption that every gene occurs exactly once in each considered genome. To allow for duplications and deletions, a relaxation to sequences of genes is necessary. A convenient way to account for the orientation of a gene within the genome is to use signed permutations or signed sequences, respectively.
Evolutionary processes can rearrange a gene order. The gene composition of some regions, however, is preserved and can be found in several related genomes. These segments, denoted as gene clusters, often contain functionally or evolutionarily associated genes (Lawrence and Roth, 1996; Overbeek et al., 1999). Hence, the analysis of gene clusters can give clues about the function of genes and valuable insights into evolutionary processes like rearrangement processes or lateral gene transfer. Various formal definitions of gene clusters based on different models of gene order have been discussed and analyzed in the literature. For surveys of different concepts of gene clusters, see Hoberman and Durand (2005) and Bergeron et al. (2008). Whenever the genomes of several species comprise the same gene cluster, it was presumably inherited from a common ancestor. Recent studies (Bergeron et al., 2004; Adam et al., 2007; Chauve and Tannier, 2008; Stoye and Wittler, 2009) build on this idea to reconstruct ancient gene clusters and to infer ancient gene orders. More precisely, the internal nodes of a given phylogenetic tree are labeled with sets of gene clusters, based on the gene orders of contemporary species at the leaves of the tree. Besides the pure identification of gene clusters, such reconstructed scenarios for the origin of the clusters and the development of the gene order can give valuable information about underlying evolutionary processes, the ancestral history of the species, and functional and evolutionary relations of genes.
Proposed reconstruction approaches differ in the underlying models for gene order and gene clusters, and in the applied methodology. However, a general aim is to ensure consistency: For a set of putative ancient gene clusters, there should exist at least one gene order that comprises all given clusters. Otherwise, the reconstruction result would be inconsistent with respect to the genome model.
The goal of reconstructing consistent labelings was first introduced by Bergeron et al. (2004) who presented an algorithm that reconstructs sets of framed common intervals on permutations. Adam et al. (2007) applied the parsimony principle as an objective function to reconstruct common intervals on permutations. A heuristic is used to reach consistency. Recently, Chauve and Tannier (2008) proposed a methodology to reconstruct the gene order of the amniote genome, based on consistent labelings of common intervals and adjacencies. In our previous work (Stoye and Wittler, 2009), we introduced an algorithmical framework that is not restricted to a specific model but instead follows an oracle-based approach to compute most parsimonious consistent labelings for various models.
All of the above methods have been successfully applied to real data and proven to yield reasonable and valuable results. They all rely on permutation-based models, which enable efficient algorithms and data structures. In particular, the verification of consistency can be translated to the well-studied Consecutive Ones Problem and be solved in polynomial time and space using data structures like PQ-trees or PC-trees (Booth and Lueker, 1976; Habib et al., 2000; Hsu, 2002; Hsu and McConnell, 2004). Some reconstruction approaches could be easily adapted to the model of sequences without duplications which allows genes to be missing in some genomes but still requires each gene to occur at most once in each genome.
In this article, we discuss consistency for sequence-based gene cluster models. Particularly, we consider the simple model of adjacencies, the classical model of common intervals (Uno and Yagiura, 2000), and two variants of the latter. For each of these models we address the problem: Given a set of gene clusters and a maximum copy number for each gene, decide whether there exists a valid gene order that contains all of the clusters. Our results range from algorithms that verify consistency for adjacencies in linear time to the confirmation of NP-completeness for the more complex models.
The present article extends a conference version presented at the RECOMB satellite workshop on Comparative Genomics (Wittler and Stoye, 2010). Here, we present much stronger NP-hardness results, emphasizing the strict complexity border between adjacencies and only slightly more relaxed models. In fact, for two of the models discussed, we rule out fixed-parameter-tractability in any natural parameter, and for a third model, only a small gap remains.
This article has been organized in the following way. First, we formally introduce the Consistency Problem in Section 2. Then, in Section 3, we give an efficient solution for the gene cluster model of both signed and unsigned adjacencies. In Section 4, we present NP-completeness results for the model of common intervals and its variants, before we finish with some discussions and conclusions in Section 5.
2. The Consistency Problem
Assume a set of putative gene clusters, assigned to an ancestral node in a given tree. These ancient clusters in turn imply a set of putative ancient genomes: all those which contain all of the given clusters. Depending on the gene cluster model used, this set of genomes can be empty if some of the clusters derived from different contemporary species are in contradiction with others. For example, when we model gene order as permutations, there is no valid gene order comprising the three adjacencies {a, b}, {a, c} and {a, d}, because, according to the model, gene a can only occur once and thus only be the neighbor of two other genes.
In a more general case, we represent genomes as sequences of genes or other genomic markers. In a sequence, any element can occur multiple times or not at all, which, in the context of gene order comparison, corresponds to paralogous genes and gene deletions, respectively.
If we allow each gene to appear arbitrarily often in any genome, the question of consistency would become redundant: Any set of gene clusters is consistent since there is a valid gene order containing all assigned clusters. For instance, we can simply create a short sequence of genes according to each cluster separately and then concatenate these sequences to an absurd yet valid gene order. Such a construction is possible for any gene cluster model. As a consequence, consistency always holds and does not contribute to a specification of reasonable reconstruction results.
Even if we replace the naive concatenation approach and instead construct preferably compact valid gene orders, we cannot avoid to include some genes multiple times. In some cases, this causes side effects. In the example given in Figure 1, the classical parsimony principle is applied to assign gene clusters to the inner nodes of a given tree, minimizing the number of gains and losses of clusters. Although a gene is contained in all input genomes only once, it is reconstructed to occur multiple times for ancestral nodes. In this simple example, we consider a subsequence of only three genes in each input genome and obtain a segment of five genes for the examined internal node. In general, such artifacts imply unnaturally long genomes for higher levels in the tree.
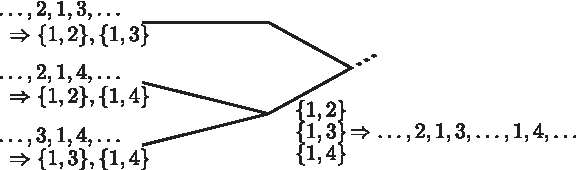
Example for an artifact that arises when gene clusters are reconstructed on the basis of gene orders, where any gene can occur arbitrarily often. A small subtree over three genomes is shown exemplarily. The gene orders on the leaves imply, beside others, the listed adjacencies. Any most parsimonious labeling would assign all three adjacencies to the lowermost internal node, implying at least two copies of gene 1. Since genes are allowed to appear multiple times in a genome, valid gene orders exist, all of which contain gene 1 at least two times—for instance the given gene order.
To preclude this unwanted effect, we refine the concept of consistency. Instead of simply restricting the total length of a genome, we limit the multiplicity of each individual gene.
In the following problem definition, we intentionally refrain from specifying a concrete model of gene clusters and instead use the imprecise notion of a sequence containing a cluster. For instance, in the simple model of gene adjacencies, a sequence g contains a gene cluster {a, b} if and only if the genes a and b occur adjacently in g.
Definition 1 (Consistency Problem)
Let (i) s contains each gene g at most m(g) times, and (ii) s contains all gene clusters
Whenever we want to consider consistency as a reconstruction criterion, we have to provide a solution for the above problem. As we will see in the following sections, the problem complexity highly depends on the specific cluster definition.
In our framework, we assume that the gene multiplicities are given. Nevertheless, we want to sketch some ways to specify m(g) for the internal nodes in the phylogenetic tree.
The threshold ml for a leaf l is obviously given by the input: We simply count the occurrences of each gene g in the gene order assigned to l to define ml(g). However, we have to determine mv for the internal nodes.
For some specific datasets, we can rely on knowledge about the genomic history. For instance, several studies suggest two whole genome duplications in the evolution of the Chordate genome in the teleost fishes lineage (Jaillon et al., 2004). Such information can be used to deduce the ancestral number of genes.
Otherwise, the most accurate but also elaborate approach would be to deploy gene-tree species-tree reconciliation (Page and Charleston, 1997) to reconstruct the history of the genes in terms of speciation events, gene duplications and gene losses. While less extensive and more suitable for our needs, one could also utilize approaches which do not require any further data or pre-knowledge. Probability-based methods (Csűrös and Miklós, 2009) could be applied to effectively and reliably infer ancestral gene multiplicities mv(g) for all internal nodes v, given the copy number at the leaves. Or, we could apply the concept of parsimony and minimize the amount of copy number differences. A less restrictive solution is to define the multiplicity of a gene g for node u in a bottom-up fashion as the maximum over the multiplicities of its child nodes
Instead of performing a separate preprocessing step to fix the thresholds in advance, one could also try to include the gene multiplicity into the overall objective of the reconstruction. However, in general, optimizing for a combination including an original objective, consistency, and the gene copy number would be an intricate task due to the strong interdependencies of the sub-criteria.
3. An Efficient Solution for Adjacencies
Probably the simplest formalization of co-localization of genes is the concept of adjacencies, i.e., pairs of directly neighboring genes. This elementary pattern of gene order conservation, also known as gene pairs or neighboring genes, has been widely used in whole genome comparison. Especially in the field of gene order reconstruction, this model is one of the most prevalent concepts (Ma et al., 2006; Bhutkar et al., 2007; Chauve and Tannier, 2008).
3.1. Unsigned adjacencies
In the following, we formalize the concept of adjacencies and present a method to efficiently solve the consistency problem for adjacencies on sequences, i.e., to decide if there exists a sequence that contains a set of given adjacencies while considering each gene g at most m(g) times. To model the problem, we use a graph theoretic approach.
Definition 2 (Unsigned Adjacencies on Sequences)
Let
Definition 3 (Gene Order Graph)
Let
The gene order graph of a set of adjacencies C can be constructed in
Lemma 1
Let (i) deg(vg) ≤ 2m(g) for all vertices (ii)
Proof
Assume we have given
If condition (i) of the lemma holds, then all nodes in the obtained extended graph have even degree: All vertices vg ≠ v0 are filled up to a degree of 2m(g) and v0 is incident to
Conditions (i) and (ii) imply that HN is Eulerian. That means, there is a closed walk (Eulerian cycle) which contains all edges, especially the edges of the original gene order graph, exactly once. Since each node vg ≠ v0 has a degree of 2m(g), it is traversed exactly m(g) times. Each such Eulerian path corresponds to a sequence of genes that contains all adjacencies in C and each gene g exactly m(g) times, as exemplified in Figure 2. Thus, C is consistent with respect to m.
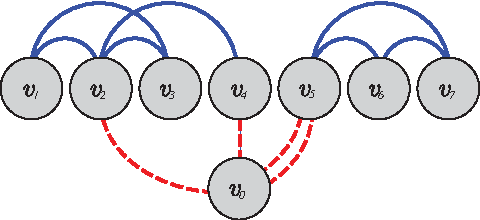
An example to illustrate the proof of Lemma 1. Consider the set of genes
On the contrary, if condition (i) is not satisfied, there is at least one gene g that is contained in more given adjacencies than its multiplicity m(g) allows. And, if condition (ii) does not hold for any connected component c, the maximum number of adjacencies of all genes in c is exhausted and the genes cannot be put into a linear order, i.e., a cycle containing v0, with the remaining genes. In both cases, the existence of a valid gene order is precluded and, thus, consistency is disproven. ▪
3.2. Signed adjacencies
A slightly more sophisticated variant of the adjacency model is motivated by the observation that the orientation of genes can play a role in co-expression and also in gene order conservation (Huynen et al., 2001).
Definition 4 (Signed Adjacencies on Signed Sequences)
Let
Note that the representation of a signed adjacency as an unordered pair is accurate since the definition of containedness does not depend on the actual assignment of a and b.
Example 1
Consider the model of signed adjacencies for N = 4. The signed adjacency {2, −3} is contained in both sequences s1 = (1, 2, 3, 4) and s2 = (4, 1, −3, −2). No other signed adjacencies of the genes 2 and 3 are contained in any of the two sequences.
We transfer the general idea from the unsigned to the signed case. To this end, we adjust the definition of the gene order graph. Now, each gene g is represented by two nodes in the graph, where each such pair is connected by m(g) edges.
Definition 5 (Signed Gene Order Graph)
Let
Similarly to the unsigned case, we can construct the graph in
Lemma 2
Let (i) deg(vg) ≤ 2m(|g|) for all vertices (ii)
Proof
We proceed analogously to the unsigned case described in the proof of Lemma 1. We extend the signed gene order graph
Then, again, the conditions (i) and (ii) of Lemma 2 imply the existence of an Eulerian path in
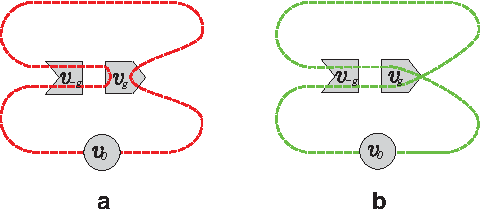
Illustration of the relation of improper
Based on the definition of a gene order graph, Lemmas 1 and 2 provide algorithms to solve the consistency problem on adjacencies on sequences in time and space linear in the number of genes and in the number of given adjacencies. Both the models and the lemmas can easily be modified to allow one circular gene order or even several circular chromosomes. Only the connectivity requirement has to be relaxed correspondingly.
4. NP-Completeness for Common Intervals
To find larger conserved regions, we now address a model for gene clusters that, in contrast to adjacencies, generally spans more than two genes: common intervals, segments of the genome containing the same set of genes in an arbitrary order but not interrupted by other genes.
The term interval stems from the original, mathematical problem statement. There, a common interval is defined on a set of permutations which is, without loss of generality, assumed to include the identity
The detection of common intervals conserved among several gene orders is a well studied problem. For details, we refer to the recent review of Bergeron et al. (2008).
As already detailed in Introduction, common intervals were successfully applied in ancestral gene order reconstruction (Adam et al., 2007; Chauve and Tannier, 2008; Stoye and Wittler, 2009).
4.1. Basic common intervals
In line with other studies, we base our definition on the notion of character sets, which enables us to formalize the cluster model in a straightforward way. Since we utilize this term for models on signed sequences later on, we directly define it for the general, signed case. Although, in our framework, a common interval is defined on a single gene order, we stick to the term common to not confuse the reader familiar with this gene cluster model by redefining the same concept under a different name.
Definition 6 (Character Set)
Let
Definition 7 (Common Intervals on Sequences)
Let
A common interval can occur multiple times in one genome. Furthermore, one occurrence of a common interval in a genome may contain several occurrences of the same gene, as illustrated by the following example.
Example 2
Consider the above model for N = 6 and sequence s = (5, 4, 2, 1, 2, 3, 6). Then, the common interval {1, 2, 3, 4} is contained in s as illustrated below, where the corresponding substring is underlined:
Recall that we want to find an answer to the question: Given a set of common intervals C and a multiplicity threshold function m, is there a valid gene order that contains all elements of C and meets the restrictions imposed by m? As we will show now, this problem is NP-complete.
Theorem 1
The consistency problem for common intervals on sequences is NP-complete, even if max{m(g)} = 2 and
Before giving the proof, we would like to emphasize that this is the strongest possible result. If the maximum multiplicity would be one, the problem becomes the polynomially solvable Consecutive Ones Problem (Booth and Lueker, 1976). If the maximum cluster size is restricted to two, this corresponds to the model of adjacencies, for which we gave a polynomial algorithm in the previous section.
Proof
One can easily formulate an algorithm that verifies a given solution, i.e., a proper gene order, for correctness in polynomial time, which shows that the problem belongs to the complexity class NP.
We will show NP-hardness of the consistency problem on common intervals by reduction from 3SAT(3), which has been proven to be NP-complete by Papadimitriou (1994). 3SAT(3) is a restricted version of 3SAT in which every variable has exactly two positive and one negative occurrence in the clauses.1 The general technique of the reduction is similar to that used in Maňuch and Patterson (2010) to prove NP-hardness for a generalized variant of the Consecutive Ones Problem. Please note that, in the rather abstract ambience of this proof, we will use the term object instead of gene.
Given a 3SAT(3) formula ϕ with variables
For this instance ϕ of 3SAT(3), we say that a clause selects one of its literals in a truth assignment of ϕ if this literal has value true in this assignment. Obviously, a truth assignment of ϕ is a satisfying truth assignment if and only if every clause selects at least one literal and for every
For each 2-clause ci with literals
For each 3-clause ci with literals
Figure 4 shows graphical representations of these gadgets, which also highlights that an instance of the consistency problem for common intervals on sequences can be viewed as a hypergraph with a vertex for each object and a hyperedge for each common interval. A sequence that is consistent with this instance is then a collection of walks on this hypergraph that covers each hyperedge, that is, for each hyperedge e there is a connected subwalk containing all and only vertices in e, such that no vertex v is visited more than m(v) times.
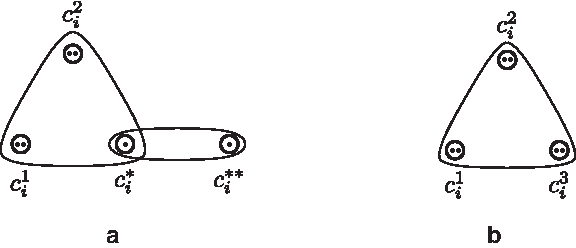
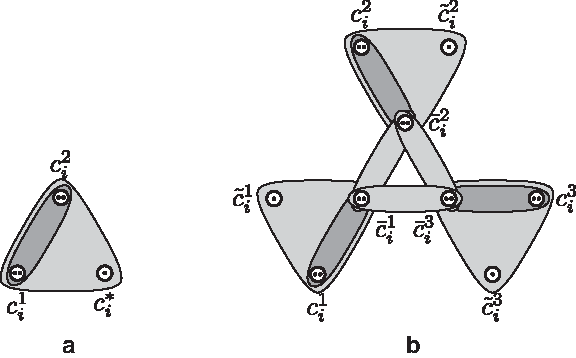
Graphical representations of the 2-clause gadget
We say that a literal object
Now, all 3n literal objects
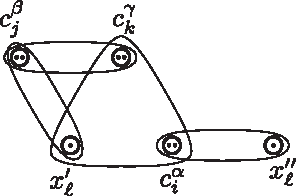
Graphical representation of the variable gadget for variable xℓ with positive occurrences
We will show that the variable gadget for xℓ ensures that in a valid string literal
We now show that the converse holds, namely if ϕ has a satisfying truth assignment τ, then
For each clause ci, we pick one literal 1. if τ(xℓ) = false, we create the substrings 2. if τ(xℓ) = true, we create the substring
The requirements imposed by all given common intervals are fulfilled. It remains to be shown that the substrings can be merged to one string s that satisfies the multiplicity restrictions as well. First, we should note that each of the objects with multiplicity one,
Consider a literal object o that appears twice in the variable substrings. It is either
Eventually, any concatenation of the remaining substrings yields a string s that is consistent with
Since the number of objects used in the construction is at most 5n + 2m, the number of common intervals is at most 4n + 2m, and each common interval is of size at most three, i.e., the construction is linear in the size of ϕ, it can be built in linear time, and hence the consistency problem on common intervals is NP-hard. ▪
Besides its classical definition, there are different generalizations of common intervals on sequences discussed in the literature, such as r-window clusters (Friedman and Hughes, 2001; Durand and Sankoff, 2002), and max-gap clusters (He and Goldwasser, 2005; Hoberman and Durand, 2005; Pasek et al., 2005), or approximate gene clusters (Rahmann and Klau, 2006; Böcker et al., 2009). Since the consistency problem is NP-complete for basic common intervals, any generalization is NP-hard as well.
In contrast to generalizations, there are also other cluster models which are restricted variants of common intervals. In the following, we will discuss such models, in particular framed and nested common intervals.
4.2. Framed common intervals
This gene cluster model, common intervals framed by two genes whose orientations have to be conserved, was first introduced on permutations as conserved intervals (Bergeron and Stoye, 2006). In gene order reconstruction, framed common intervals on permutations was the first model to formally state the problem of finding putative ancestral sets of gene clusters preserving consistency (Bergeron et al., 2004).
Definition 8 (Framed Common Intervals on Signed Sequences)
Let
According to this definition, a gene can be an extremity and an inner element, or even a left and right extremity at the same time. Apart from that, analogously to basic common intervals, a cluster can occur multiple times in one genome, and one gene can be contained several times in one cluster occurrence, as illustrated by the following example.
Example 3
Consider the model of framed common intervals for N = 6 and sequence s = (5, 4, −2, −1, 2, −3, 6). Besides others, the framed common interval [4{1, 2} −3], is contained in s as illustrated by the box diagram below, where the occurrences of the extremities and the inner elements are surrounded by rectangles:
The obvious relationship of basic and framed common intervals allows us to infer an important correlation of these models with respect to the consistency problem: Any instance of this problem for common intervals can be reduced to an instance of framed common intervals. Based on this, we can deduce the following statement.
Theorem 2
The consistency problem for framed common intervals on signed sequences is NP-complete, even if max{m(g)} = 2 and
Proof
Again, one can easily formulate an algorithm that verifies a given solution for correctness in polynomial time, which shows that the problem belongs to the complexity class NP.
NP-hardness is shown by reducing the basic common intervals used in the proof of Theorem 1 to framed common intervals.
The basic idea is to replace each common interval
Since the given basic common intervals used in the proof of Theorem 1 overlap, the framing elements have to be included into the set of inner elements of overlapping intervals. We use the following technique to ensure that, if there is a valid gene order for the basic common intervals, there is a valid gene order for the constructed set of framed common intervals. Together with the argument in the previous paragraph, this will yield equivalence of the two instances of the consistency problem.
For each basic common interval
What remains to be shown is that such a construction is possible using at most six inner elements in each framed common interval, as well as that a maximum multiplicity of two is sufficient.
To minimize the number of inner elements, we do not always add both framing elements to all overlapping intervals. The structure of the common intervals used in the gadgets of the proof of Theorem 1 restricts the possible overlaps of their occurrences in a valid gene order. As can be seen in the proof of Theorem 1, if there is a valid sequence, we can construct one using the following orders (or their reversals) of interval occurrences within the gadgets:
Within the gadget,
A 2-clause gadget ci overlaps the gadgets of two variables, say xj and xk. As can be seen in the proof of Theorem 1, if there is a valid sequence, we can construct one with one of the following orders (or their reversals) of interval occurrences:
where
A 3-clause gadget ci overlaps the gadgets of three variables, say xj, xk and xℓ in the element
where
In summary, we reduce a given set of common intervals as used in the proof of Theorem 1 to a set of framed common intervals with at most six inner elements as follows:
For the basic common intervals
For the basic common intervals Si1,2 used in the 2-clause gadget for c
i
, we create the framed common intervals
where we define
For the basic common interval Si used as in the 3-clause gadget for ci, we create the framed common interval
where we define
It remains to be shown that a maximum multiplicity of two for all newly added elements suffices. This is true, because each new element is included in the inner elements of at most two intervals. In fact, we can assign a multiplicity of one to some of the objects. We define:
Since the number of objects used in the construction is at most 6n + 8m, the number of framed common intervals is at most 4n + 2m, and each framed common interval contains at most six inner elements, i.e., the construction is linear in the size of ϕ, it can be built in linear time, and hence the consistency problem on common intervals is NP-hard. ▪
Please note that, again, for a maximum multiplicity of one, polynomial solutions exist. Framed common intervals with no inner elements are equivalent to signed adjacencies, for which we gave an efficient solution. However, there is a gap left for framed common intervals with one to five inner elements. For these, the complexity is still open.
4.3. Nested common intervals
Hoberman and Durand (2005) discussed nestedness as a desired property of gene clusters and proposed a first algorithm to identify respective clusters. Recently, nested common intervals were formally defined and studied in Blin et al. (2010), and gave efficient algorithms to detect them in genomes modeled both as permutations and as sequences.
Definition 9 (Nested Common Intervals on Sequences)
Let (i) an unordered pair of genes {a, b} with a ≠ b, which is contained in a sequence s over (ii) an unordered pair {c, a} of a nested common interval c and a gene a, which is contained in a sequence s if and only if, in s, a is adjacent to a substring s′ of s such that
where the character set of a nested common interval is the set of all contained genes:
Similar to the other cluster models discussed above, any nested common interval may occur multiple times in one genome and one gene may be contained multiple times in the occurrence of a cluster in one genome. Analogously to framed common intervals, one gene may be incorporated in the definition of one cluster several times.
Example 4
Consider the model of nested common intervals for N = 6 and sequence s = (5, 4, 2, 1, 2, 3, 6). Then, besides others, the nested common interval {{{2, 3}, 1}, 4} is contained in s as illustrated below, where the occurrences of the subclusters are indicated by lines:
In contrast, {{{1, 3}, 2}, 4} is not contained in s since, although gene 4 is adjacent to a substring with character set {1, 2, 3}, none of the occurrences of gene 2 is adjacent to a substring with character set {1, 3}.
Note that cluster {{2, 3}, 3} is not contained in g, because 3 is not adjacent to a substring with character set {2, 3}, whereas cluster {{1, 2}, 2} is contained in s:
Even the strict assumption of nestedness is not strong enough to allow an efficient verification of consistency. In fact, similar to basic common intervals, there is no gap left for fixed-parameter tractability in the considered parameters.
Theorem 3
The consistency problem for nested common intervals on sequences is NP-complete, even if max{m(g)} = 2 and
Proof
NP-hardness is proven by reduction from 3SAT(3) using a construction very similar to that of Theorem 1. Given 3SAT(3) formula ϕ, we will again design an instance
For each 2-clause ci with literals
Graphical representations of the 2-clause gadget
For each 3-clause ci with literals
Note again that in both clause gadgets, at least one of the literal objects is selected in any valid string s. For the 2-clause gadget, string s has to contain one of the substrings
For each variable xℓ, we will use the same construction as in the proof of Theorem 1 with one exception that instead of the basic common interval
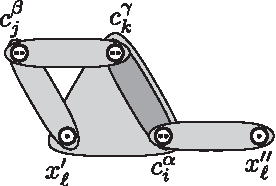
Graphical representation of the variable gadget for variable xℓ with positive occurrences
We now show that the converse holds, namely if ϕ has a satisfying truth assignment τ, then
For each clause ci, we pick one literal
It can be easily checked that the requirements imposed by all given nested common intervals are fulfilled. It follows by the same argument as in the proof of Theorem 1 that the created substrings can be merged and concatenated into a valid string s. Thus, if ϕ has a satisfying assignment τ, then
Since the number of objects used in this construction is at most 5n + 6m, the number of nested common intervals is at most 5n + 9m, and each nested common interval is of size at most three, i.e., the construction is linear in the size of ϕ, it can be built in linear time, and hence the consistency problem on common intervals is NP-hard. ▪
4.4. Further variations and restrictions
Our NP-completeness results also hold for further variations of the above models.
If we preclude a nested common interval to contain any gene multiple times, e.g., {{a, b}, a}, or if we preclude any gene to be left and right extremity of a framed common interval, e.g., [a I a], our proof techniques still apply and thus NP-completeness still holds. Also, if we restrict any occurrence of a common or nested common interval within a genome to contain each gene only once, NP-completeness still holds.
Instead of restricting the multiplicity for each gene individually, one could define a maximum total number of genes, i.e., a maximum genome length. But going back to the NP-hardness proofs, we find that they also hold in this model. In the proofs for basic and nested common intervals, each gene g with multiplicity m(g) occurs in at least 2m(g) − 1 intersecting gene clusters such that all “allowed” copies of that gene are actually required. Thus, there is no flexibility left to use one gene only m(g1) − 1 times and another gene m(g2) + 1 times. Also, the auxiliary genes used in the proof for framed common intervals have all to be used exactly as often as specified. Hence, in all models, the proofs also hold if only
Since a typical prokaryotic genome consists of one circular chromosome, we are also interested in modeling gene order as circular sequences. In fact, based on the above NP-completeness results, we can deduce NP-completeness of the consistency problem for all the considered gene cluster models on circular sequences in a straight forward fashion.
To redefine the cluster models, we allow any gene cluster to appear at the end of a sequence
Corollary 1
The consistency problem on circular sequences is NP-complete for the gene cluster models of basic, framed and nested common intervals for the maximum multiplicities and cluster sizes as stated in Theorems 1–3.
Proof
One can easily formulate an algorithm that verifies a given solution for correctness in polynomial time, which shows that the problem belongs to the complexity class NP.
The consistency problem for the considered gene cluster models on linear sequences is NP-complete according to Theorems 1, 2 and 3.
Let the set of genes • • C′:= C ∪ {c}, where c is a gene cluster containing exactly the genes N + 1 and N + 2 (the basic or nested common interval {N + 1,N + 2}, or the framed common interval [N + 1{}N + 2], respectively), •
If C is consistent with respect to m, then there is a linear sequence
Now, we show the opposite implication: If C′ is consistent with respect to m′, then there is a circular sequence
So far, we only considered genomes composed of one chromosome. As already mentioned in Section 3, the algorithm presented for adjacencies can easily be modified to handle several chromosomes—linear or circular. Also, the NP-completeness results hold for several linear chromosomes. In fact, the proofs are based on the construction of several substrings which could be kept as separate chromosomes instead of concatenating them to one string. In contrast, the proof of the latter lemma builds on exactly one circular chromosome. It is open whether NP-completeness holds for the case of several circular chromosomes for these models.
5. Conclusion
In this article, we have discussed the consistency problem, i.e., the problem of deciding whether there exists a valid gene order comprising a given set of gene clusters. We have discussed this question for different gene cluster models on sequences with restricted gene multiplicities. In summary, we identified a strict border between gene cluster models for which we can verify consistency efficiently and those for which we cannot. The complexity rises drastically from linear time for adjacencies to NP-hard for more general cluster models, even if they are strongly restricted.
This raises the question for a sequence-based gene cluster model that, on the one hand, allows some degree of flexibility and, on the other hand, offers a polynomial-time algorithm to verify consistency. The integration of such a model into any of the existing reconstruction methods could increase sensitivity. Actually, first results on both simulated and real data indicate that within segments of conserved gene content, the order of the genes is conserved almost exactly (Wittler, 2010). Thus, a model covering only single missing or additional genes, or the reversal of two neighboring genes could already enhance reconstruction results strongly.
On permutations, the consistency problem for common intervals is the consecutive ones property (C1P) problem (Booth and Lueker, 1976; Habib et al., 2000; Hsu, 2002; Hsu and McConnell, 2004). Thus, one approach that includes additional or missing genes is to relax the condition of the consecutivity of the ones of each row, by allowing gaps, with some restriction on the nature of these gaps. The question is then to decide if there is an ordering of the columns that satisfies these relaxed C1P conditions. In Goldberg et al. (1995), the authors introduced the k-consecutive-ones property (k-C1P): Decide if the columns of a binary matrix can be permuted such that each row contains at most k blocks. This problem is NP-complete for every k ≥ 2 (Goldberg et al., 1995), and also minimizing the number of gaps in the entire matrix is NP-complete even if each row of this matrix has at most two ones (Haddadi, 2002).
In the spirit of this approach, Chauve et al. (2009) define the gapped C1P: given two integers k and δ, a binary matrix M has the (k, δ)-C1P if its columns can be permuted such that each row contains at most k blocks and no gap larger than δ. While they show that such a property can be decided in polynomial-time when the number of ones in each row of M is bounded, they show that the general case is hard. In particular, they show for all k ≥ 2,δ ≥ 1, (k, δ) ≠ (2, 1) that the (k, δ)-C1P is NP-complete. So indeed, in this case, aside from the single open case of the complexity of the (2,1)-C1P, there is also a strict complexity border between the classical C1P ((1,0)-C1P in the gapped C1P context) and this relaxed model.
In Maňuch and Patterson (2010), the authors show also for binary matrices of bounded degree d that the k-C1P is NP-complete even when d = 3, which is quite suprising, as this is the weakest form of consecutivity requirement: in each row, only two of the ones must be adjacent. This is shown with an NP-completeness construction based on finding a collection of walks on a hypergraph H that covers each hyperedge in H, a technique that inspired some of the NP-completeness constructions in this work.
We implemented the gene order graph to model adjacencies on sequences and integrated this gene cluster model into our unified reconstruction framework, presented in Stoye and Wittler (2009) (available at bibiserv.techfak.uni-bielefeld.de/rococo/). An elaborate description of the method and the results can be found in Wittler (2010). We refrain from reporting detailed results here because these are concerned more with the reconstruction method than with the general concept of consistency discussed in this article. Nevertheless, we would like to mention the following overall findings. Simulations showed that estimating the gene multiplicities using the simple maximum approach does not significantly decrease the accuracy of the reconstruction compared to using the “real” simulated copy numbers. Furthermore, we applied our method to genomic data of Corynebacteria using different gene cluster models: Common intervals on permutations and adjacencies on sequences. A comparison of the results revealed a large overlap. Nevertheless, many conserved segments could only be identified by either of the approaches. This highlights the importance of studying gene cluster reconstruction with respect to different, especially flexible, models for gene clusters and the relaxed model of sequences for gene order.
Footnotes
Acknowledgments
We wish to thank Cedric Chauve for valuable discussions. The work of Roland Wittler was supported by the DFG Graduiertenkolleg Bioinformatik (GK 635). The work of Ján Maňuch was supported by NSERC DG. The work of Murray Patterson was supported by NSERC PGS-D3.
Disclosure Statement
No competing financial interests exist.
1
We remark that the exact formulation of 3SAT(3) in Papadimitriou (1994) allows also variables with one negated and two positive occurrences, but these can easily be converted to the other type of variables by replacing them with their negations in all clauses. Clearly, this does not affect the complexity of the problem.