We prove that the genome aliquoting problem, the problem of finding a recent polyploid ancestor of a genome, with breakpoint distance can be solved in polynomial time. We propose an aliquoting algorithm that is a 2-approximation for the genome aliquoting problem with double cut and join distance, improving upon the previous best solution to this problem, Feijão and Meidanis' 4-approximation algorithm.
1. Introduction
Comparing two genomes with duplicated genes is difficult. None of the distances used to compare genomes today (e.g., breakpoint distance, reversal distance, double cut and join distance) handle duplicated genes. However, in the special case where all genes are duplicated the same number of times, there has been some success.
Informally, the genome aliquoting problem is the problem of finding a genome with one copy of every gene given a genome with exactly p copies of every gene such that the distance between the given and resulting genomes is minimized according to some distance metric. Thus, the genome aliquoting problem eliminates the duplicate genes allowing a genome to be compared with other genomes using an existing algorithm. Solving this problem will allow genomes which have undergone a recent polyploidization event, common in plants, to be compared.
There have been a number of solutions to the genome aliquoting problem where the genome has exactly two copies of every gene. This restricted version of the problem is called the genome halving problem and was first introduced in El-Mabrouk and Sankoff (2003). It was solved for reversal and translocation distance in El-Mabrouk and Sankoff (2003) and Alekseyev and Pevzner (2007), and for double cut and join distance in Warren and Sankoff (2009b) and Mixtacki (2008).
The genome aliquoting problem was introduced in Warren and Sankoff (2009a) along with a sketch of a heuristic algorithm for the problem under double cut and join distance. Feijão and Meidanis (2009) provided an exact solution under single cut or join distance which is also a 4-approximation algorithm under double cut and join distance. In this article, we provide an exact polynomial-time algorithm under breakpoint distance which is also a 2-approximation algorithm under double cut and join distance. Since our algorithm is similar to that presented in Warren and Sankoff (2009a), it also bounds that heuristic as a 2-approximation for double cut and join distance.
2. Duplicated Genomes
The fundamental elements that we study are genes. We represent each gene as a pair of extremities such that a gene x is represented by its head\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{x}$$
\end{document}, which corresponds to the 3′ end of the gene, and its tail\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\stackrel{\succ}{x}$$
\end{document}, which corresponds to the 5′ end of the gene. A genome is represented by a multiset of 2-multisets and 1-sets of extremities, called adjacencies and telomeres respectively, such that, for each gene, the genome contains exactly the same number of heads as tails. The functions \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal G} (G) \ {\rm and} \ {\cal E} (G)$$
\end{document} return the set of all genes or extremities respectively of a genome G. The collection of copies of the same gene are called a gene family with the size being the number of copies in the genome (i.e., the number of heads, or, alternatively, the number of tails, that appear in a genome).
Genomes with one or more gene families with size greater than one are challenging to manipulate. Not only must there be the same number of heads and tails, but to understand the layout of the genome, we must know for duplicated genes which head corresponds to which tail; otherwise, the layout of the genome is not unique. Similarly, to compute the distance of two genomes, we must know which copy in one genome matches which copy in another genome. Genomes where this information is known are called ordered genomes. In such genomes, we distinguish members of the same gene family by a subscript such that each head corresponds to the tail with the same subscript, e.g., \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{a_1}$$
\end{document} corresponds to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\stackrel{\succ}{a_1}$$
\end{document}. Similarly, if we know the ordering between two genomes then each extremity in one genome must correspond to the extremity with the same subscript in the other genome, e.g., \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{a_1}$$
\end{document} in one genome must correspond to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{a_1}$$
\end{document} in the other genome. By default, we assume most genomes are ordered with themselves, but the challenge of comparing genomes with duplicated genes comes from finding an ordering between two genomes as different orderings change the distance. Thus, to compare genomes with duplicated genes, we must find an ordering between them that minimizes their distance.
For most situations, ordered genomes are all that is needed. However, ordered genomes can be difficult to use, e.g., \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\vec{a_1}, \vec{b_1}\} \cap \{ \vec{a_2}, \vec{b_2}\} = \emptyset$$
\end{document}, and yet, frequently, it is desirable to perceive them as equal. Since these problems tend to occur frequently in our work, we introduce the concept of an unordered genome where no effort is made to distinguish the genes. Let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} be the unordered counterpart of an ordered genome G. Similarly, if α is an element of G, then \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{\alpha}$$
\end{document} is an element of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} and if S is a subset of G then \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{S}$$
\end{document} is a subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. Unordered extremities are distinguished from ordered extremities by the absence or presence respectively of a subscript. Transforming an ordered genome to an unordered genome is trivial but the reverse can be very difficult and is problem specific.
The key concept that helps solve the genome aliquoting problem is perfection:
Definition 1
We define perfection between two elements of a genome to be that the two elements are the same or are disjoint. For adjacencies we also have the special requirement that they are not of the form {x, x} for some extremity x. Since there are two types of elements, adjacencies and telomeres, two elements α and β can be in one of the following three configurations:
• let α and β be a pair of adjacencies. They are perfect if and only if they are disjoint or the same and neither are of the form {x, x} for some extremity x;
• let α and β be a pair of telomeres. Since all telomeres are either the same or disjoint all pairs of telomeres are perfect;
• let α be an adjacency and β be a telomere, since they cannot be the same they must be disjoint in order to be perfect. Also, α must not be of the form {x, x} for some extremity x;
A genome, or any set of adjacencies and telomeres, is perfect if and only if all pairs of elements are perfect.
For example, given a genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{\{\vec{a_1}\}, \{\stackrel{\succ}{a_1}, \stackrel{\succ}{b_1}\},\{\vec{b_1}, \stackrel{\succ}{d_1}\}, \{\vec{d_1}, \stackrel{\succ}{e_1}\}, \{\vec{e_1}, \stackrel{\succ}{b_2}\}, \{\vec{b_2} \}, \{\vec{a_2}\}, \{\stackrel{\succ}{a_2}, \vec{c_1}\},\{\stackrel{\succ}{c_1}, \stackrel{\succ}{c_2}\}, \{\vec{c_2}, \vec {d_2} \}, \{\stackrel{\succ}{d_2}, \stackrel{\succ}{e_2}\}, \{\vec{e_2}\}\}$$
\end{document} its corresponding unordered genome is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde {G} = \{\{\vec{a} \}, \{\stackrel{\succ}{a}, \stackrel{\succ}{b}\}, \{\vec{b}, \stackrel{\succ}{d}\}, \{\vec{d}, \stackrel{\succ}{e}\}, \{\vec{e}, \stackrel{\succ}{b} \}, \{\vec {b}\}, \{ \vec {a} \}, \{\stackrel{\succ}{a}, \vec{c}\}, \{\stackrel{\succ}{c}, \stackrel{\succ}{c} \}, \{\vec {c}, \vec{d}\}, \{\stackrel{\succ}{d}, \stackrel{\succ}{e}\}, \{\vec{e}\}\}$$
\end{document}. In \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}, the adjacencies \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\stackrel{\succ}{a}, \stackrel{\succ}{b}\} \ {\rm and} \ \{\vec {b}, \stackrel{\succ}{d}\}$$
\end{document} are perfect because they are disjoint but \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\stackrel{\succ}{a}, \stackrel{\succ}{b} \} \ {\rm and} \ \{\stackrel{\succ}{a}, \vec{c}\}$$
\end{document} are not perfect because they are neither disjoint nor equal (they have \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\stackrel{\succ}{a}$$
\end{document} in common but do not share the other extremity). The adjacency \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \stackrel{\succ}{c}, \stackrel{\succ}{c}\}$$
\end{document} can never be perfect because it is of the form {x, x} for some extremity x. The telomere \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \vec{a}\}$$
\end{document} is perfect when compared with both \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \vec{a}\} \ {\rm and} \ \{ \vec{e}\}$$
\end{document} since it is equal to the former and disjoint with the latter. \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \vec{e}\}$$
\end{document} is not perfect when compared with the adjacency \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\vec {e}, \stackrel{\succ}{b}\}$$
\end{document} because they are not disjoint.
In biology, a polyploid is a genome with multiple copies of the same chromosome. A perfect unordered genome where all gene families are of the same size shares this property and, hence, is called a polyploid. Similarly, an ordered genome G whose corresponding unordered genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} is a polyploid is a polyploid.
For example, the ordered genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{\{\vec {a_1} \}, \{\stackrel{\succ}{a_1}, \stackrel{\succ}{b_1}\}, \{\vec {b_1} \}, \{ \vec {b_2} \}, \{ \stackrel{\succ}{a_2}, \stackrel{\succ}{b_2}\}, \{\vec {a_2}\} \{\vec {e_2} \}, \{\stackrel{\succ}{d_2}, \stackrel{\succ}{e_2} \}, \{\vec{c_2}, \vec{d_2}\}, \{\stackrel{\succ}{c_2}\}, \{ \vec {e_1} \}, \{\stackrel{\succ}{d_1}, \stackrel{\succ}{e_1}\}, \{\vec {c_1}, \vec {d_1} \}, \{\vec {c_1} \}\}$$
\end{document} is a polyploid because its corresponding unordered genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde {G} = \{\vec {a} \}, \{\stackrel{\succ}{a}, \stackrel{\succ}{b}\}, \{ \vec {b} \}, \{ \vec {b} \}, \{\stackrel{\succ}{a}, \stackrel{\succ}{b} \}, \{\vec {a} \} \{ \vec {e} \}, \{\stackrel{\succ}{d}, \stackrel{\succ}{e} \}, \{ \vec {c}, \vec {d} \}, \{\stackrel{\succ}{c}\}, \{ \vec{e} \}, \{\stackrel{\succ}{d}, \stackrel{\succ}{e} \}, \{ \vec {c}, \vec {d} \}, \{ \vec {c} \}$$
\end{document} is perfect and all gene families are of the same size (there are two genes in each family).
Our problem is as follows:
Definition 2
Given an ordered genome G with all gene families of size p, the genome aliquoting problem is to find a polyploid H with all gene families of size p ordered in relation to G such that the distance between G and H is minimal.
3. Breakpoint Distance
A breakpoint (BP) is a difference in adjacencies or telomeres between two genomes, e.g., in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{\{\vec {a_1} \}, \{\stackrel{\succ}{a_1}, \stackrel{\succ}{b_1} \}, \{ \vec {b_1}, \stackrel {\succ}{d_1}\}, \{ \vec {d_1}, \stackrel {\succ}{e_1} \}, \{ \vec {e_1}, \stackrel{\succ}{b_2}\}, \{ \vec {b_2} \}, \{ \vec {a_2} \}, \{ \stackrel{\succ}{a_2}, \vec {c_1} \}, \{ \stackrel{\succ}{c_1}, \stackrel{\succ}{c_2}\}, \{\vec {c_2}, \vec {d_2} \}, \{\stackrel {\succ}{d_2}, \stackrel{\succ}{e_2} \}, \{\vec {e_2} \} \} \ {\rm and} \ H = \{ \{ \vec {a_1} \}, \{ \stackrel{\succ}{a_1}, \vec {b_1} \}, \{ \stackrel {\succ} {b_1}, \vec {c_1} \},\{ \stackrel{\succ} {c_1}, \vec {d_1} \}, \{ \stackrel {\succ} {d_1}, \vec {e_1} \}, \{ \stackrel {\succ} {e_1},\vec {a_2} \}, \{ \stackrel {\succ}{a_2}, \vec {b_2} \}, \{ \stackrel {\succ} {b_2}, \vec {c_2} \}, \{ \stackrel {\succ} {c_2}, \vec {d_2} \}, \{ \stackrel{\succ} {d_2}, \vec {e_2} \}, \{ \vec {e_2} \} \}, \{ \vec {b_1}, \vec {d_1} \}$$
\end{document} is a breakpoint in G since there is no equivalent element in H but \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \vec{a_1}\}$$
\end{document} is not a breakpoint because it is in both genomes. Extending this notion to entire genomes we arrive at the following definition:
Definition 3
The breakpoint distance, introduced in Sankoff and Blanchette (1997), between two genomes G and H is the number of breakpoints between G and H.
The breakpoint distance is easy to calculate. From Tannier et al. (2009), given two genomes G and H, let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal A} (G, H)$$
\end{document} be the set of adjacencies shared between G and H and let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal T} (G, H)$$
\end{document} be the set of telomeres then the breakpoint distance between G and H can be calculated using the following equation:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{\cal D} _ {BP} (G, H) = | {\cal G} (G) | - | {\cal A} (G, H) | - \frac {| {\cal T} (G, H) |} {2} \tag {1}
\end{align*}
\end{document}
Since polyploids are perfect genomes where every gene family has the same size, our strategy to aliquote a genome G is to find a perfect subset of G and then transform it into a perfect genome. Because manipulating G directly is challenging, we will manipulate its unordered counterpart \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} instead.
There are many possible perfect subsets of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} and we need to choose the one which will minimize the distance. From Equation 1, we can see that the distance is minimized by maximizing the number of adjacencies and telomeres present in both genomes. Thus, we define the weight of a perfect set P in relation to a genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} to be:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{\cal W} (P) = {\cal A} (\tilde {G}, P) + \frac{{\cal T}(\tilde {G}, P)}{2} \tag {2}
\end{align*}
\end{document}
We define a maximum perfect set P to be a perfect subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} with maximum weight.
While it is impossible to compute the distance using an unordered genome, it is easy to see that, once transformed into a genome and reordered, a maximum perfect subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} will minimize the distance. However, not all maximum perfect sets can be transformed into an ordered genome that can be compared with G. In addition to being ordered, to be compared two genomes must have the same set of extremities and it is possible to construct a maximum perfect set that is missing extremities from \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. Thus, we introduce the notion of a covering set. A set C covers an unordered genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} if and only if it contains at least one copy of every extremity in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Unfortunately, it is impossible to always find a maximum perfect and covering subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}; for some unordered genomes no such subset exists. To solve the problem, we simply remove the restriction that it must be a subset, i.e., the maximum perfect and covering set may have elements not in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. However, without the restriction that the perfect set has to be a subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}, the idea of a maximum perfect set does not make any sense; we could add an infinite number of adjacencies and telomeres to the set if they do not have to come from the genome. Thus, we introduce a quasi maximum perfect set: a perfect set with a maximum perfect set as a subset but with additional elements not from \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. All maximum perfect sets are also quasi maximum perfect sets, although the reverse is not true.
It is easy to transform a quasi maximum perfect and covering set with respect to a genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} into a polyploid that, if ordered, could be compared with G: we simply increase the number of copies of each element. Algorithm 1 does exactly this; its correctness follows from the following lemma:
Replicate
Input:P quasi maximum perfect and covering set with respect to a genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} and p, the size of the gene families in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Output: A polyploid \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} with all gene families of size p.
4 add α to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} until the multiplicity is p
Given a genome\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}with all gene families of size p and P, a quasi maximum perfect and covering set with respect to\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}, \tilde{H} = \textsc{Replicate} (P, p)$$
\end{document}is a polyploid such that\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal E} (\tilde{H}) = {\cal E} (\tilde{G})$$
\end{document}.
Proof
There are three properties of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} that we need to prove. First, we need to prove that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is in fact an unordered genome. Second, that it is a polyploid. Third, that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal E} (\tilde{H}) = {\cal E} (\tilde{G})$$
\end{document}.
The fact that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is unordered is trivial. \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is a genome follows from the fact that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \subseteq \tilde{H}$$
\end{document} and P covers \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} and Algorithm 1 ensures an equal number of heads and tails. \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal E} (\tilde{H}) = {\cal E} (\tilde{G})$$
\end{document} also follows from this fact. Hence, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is an unordered genome and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal E} (\tilde{H}) = {\cal E} (\tilde{G})$$
\end{document}.
By definition, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is a polyploid if and only if it is perfect with all gene families of size p. From Algorithm 1, it is clear that all gene families of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} are of size p. Since P is perfect and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P \subseteq \tilde{H}$$
\end{document} and, furthermore, the only difference between P and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is the multiplicity of the elements, which does not affect the property of perfection, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} must also be perfect. ▪
5. Reorder Genes
How a genome is reordered can affect the distance. However, with a quasi maximum perfect and covering set with respect to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} as a base, it is easy to reorder the resulting polyploid such that its distance to G is minimal. Algorithm 2 reorders the genome, and the following lemma proves its correctness:
Reorder
Input: A polyploid \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} with all gene families of size p and a genome G with all gene families of size p where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal E} (\tilde{G}) = {\cal E} (\tilde{H})$$
\end{document}.
Output: A polyploid H with all gene families of size p such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H)$$
\end{document} is minimal amoung all ordered genomes of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document}.
1
H ← ∅
2
foreach element \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\alpha \in G$$
\end{document}do
foreach element \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{\alpha} \in \tilde{H}$$
\end{document}do
9
if\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{\alpha}$$
\end{document} is an adjacency then
10
let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{x, y\} = \tilde{\alpha}$$
\end{document} where x, y are extremities
11
while there exists an i such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$x_i \in E$$
\end{document} and a j such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$y_j \in E$$
\end{document}do
12
add {xi, yj} to H
13
remove xi from E
14
remove yj from E
15
end
16
else
17
let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{x\} = \tilde{\alpha}$$
\end{document} where x is an extremity
18
while there exists an i such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$x_i \in E$$
\end{document}do
19
add {xi} to H
20
remove xi from E
21
end
22
end
23
end
24
returnH
Lemma 2
Given a genome G with all gene families of size p, let P be a quasi maximum perfect and covering set with respect to\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}and let\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H} = \textsc{Replicate} (P, p)$$
\end{document}, then\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H)$$
\end{document}where\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$H = \textsc{Reorder} ({\tilde H}, G)$$
\end{document}is minimal.
Proof
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H)$$
\end{document} is minimal if there does not exist an other genome H′ such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H^{\prime}) < {\cal D}_{BP} (G, H)$$
\end{document}. Assume towards contradiction that such an H′ does exist.
Let E be the set of elements that are identical between H and G and let E′ be the set of elements that are identical between H′ and G. We observe that because H and H′ are polyploids \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E} \ {\rm and} \ \tilde{E}^{\prime}$$
\end{document} must be perfect.
From Equation 1, only adjacencies and telomeres that are the same in both genomes reduce the distance. Thus, since \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H^{\prime}) < {\cal D}_{BP} (G, H), {\cal W} (\tilde{E}^{\prime}) > {\cal W} (\tilde{E})$$
\end{document}.
By definition, a quasi maximum perfect set must have a maximum perfect set as a subset; let M ⊆ P be such a maximum perfect set of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. Since E is a subset of H, it follows that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E}$$
\end{document} must be a subset of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document}. Thus, M and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E}$$
\end{document} are subsets of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} and, in fact, we will prove that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \tilde{E}$$
\end{document} by assuming towards contradiction that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M \neq \tilde{E}$$
\end{document}.
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M \neq \tilde{E}$$
\end{document} if and only if the multiplicity of all elements in M is not equal to the multiplicity of all elements in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E}$$
\end{document}. Let α be an element that has different multiplicities in both M and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E}$$
\end{document}. Since M is a maximum perfect set of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} all of its elements can have a multiplicity of at most p and, because M is maximum, the multiplicity of α in M is equal to the multiplicity of α in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. Since Replicate(P, p) creates \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} by setting all the elements of P, which includes all of the elements of M, to multiplicity p, the multiplicity of α in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} is greater than or equal to that of M. It follows that the multiplicity of α in M is the multiplicity of α in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G} \cap \tilde{H}$$
\end{document}. From Lines 2–6 in Algorithm 2 Reorder\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$(\tilde{H}, G)$$
\end{document} we can conclude that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G} \cap \tilde{H} = (G \cap H)$$
\end{document}. By definition, E = G ∩ H, hence, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$(G \cap H) = \tilde{E}$$
\end{document}. Therefore, the multiplicities of α in M and in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{E}$$
\end{document} are equal; a contradiction. Thus, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \tilde{E}$$
\end{document}.
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal W} (\tilde{E}^{\prime}) > {\cal W} (M)$$
\end{document} follows from the facts that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \tilde{E} \ {\rm and} \ {\cal W} (\tilde{E}^{\prime}) > {\cal W} (\tilde{E})$$
\end{document}. But M is a maximum perfect set; a contradiction. Hence, H must be minimal. ▪
6. Fully Modified Clique Graphs
We have reduced the problem of aliquoting a genome to that of finding a maximum perfect and covering set in the breakpoint model. To find the maximum perfect and covering set, we construct a graph from the genome that highlights its important information.
Definition 4
A fully modified weighted clique graph of a genome\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}, denoted\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document}, is defined as follows:
• For each gene family x in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}, {\cal CG} (\tilde{G})$$
\end{document}, has four vertices, one labeled\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{x}$$
\end{document}, one labeled\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\stackrel{\succ} {x}$$
\end{document}and two without labels, called null vertices, of which one is connected to the vertex labeled with\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\vec{x}$$
\end{document}and the other to the vertex labeled with\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\stackrel{\succ}{x}$$
\end{document};
• For each adjacency {x, y} in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}where x ≠ y, there is an edge between the vertices labeled x and y with a weight equal to the multiplicity of {x, y} in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document};
• For each telomere {x} in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}, the edge between the vertex labeled x and its adjacent null vertex has a weight equal to half of the multiplicity of {x} in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document};
• All other edges in\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document}have weights of 0.
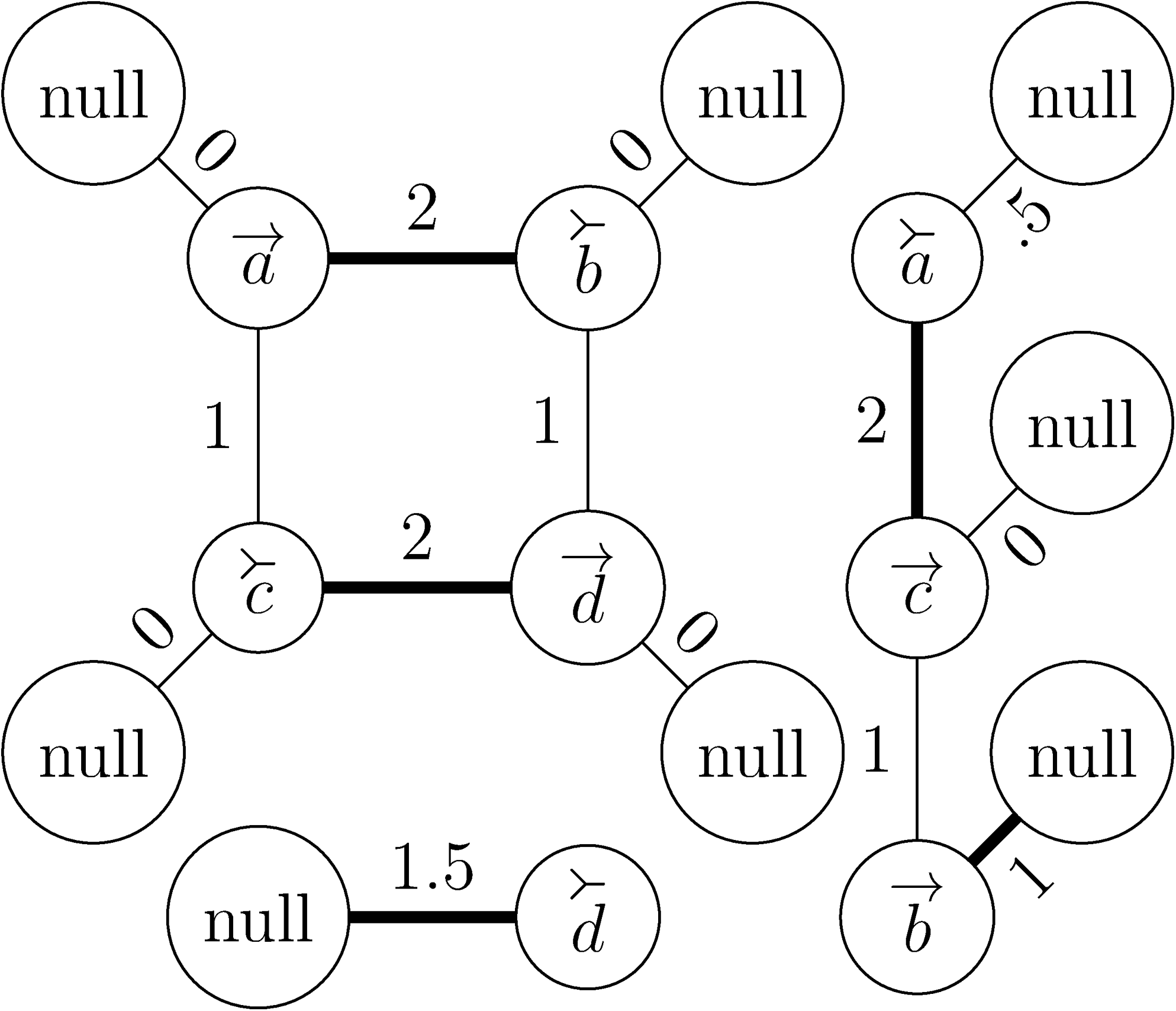
An example of a fully modified weighted clique graph. The five bold edges represent all the edges that belong to the maximum weight matching of this graph.
In the fully modified weighted clique graph, edges correspond to adjacencies or telomeres. Thus, the problem is to choose edges from the fully modified weighted clique graph such that the corresponding adjacencies and telomeres form a maximum perfect and covering set. From Definition 4, it is clear that the adjacencies or telomeres corresponding to any two edges in the fully modified weighted clique graph are perfect if and only if the edges share no vertex in common or correspond to the same edge (since there are no loops, there are no edges that correspond to adjacencies of the form {x, x} for some extremity x). Finding a set of edges that share no vertices in common is a well-studied computational problem known as the maximum matching problem, although, since our edges are weighted, in this case we are actually more interested in the maximum weight matching problem Lovász and Plummer (2009).
Given a matching, Algorithm 3 constructs its corresponding perfect set. However, it is the quality of the matching that determines the quality of the perfect set. The fully modified weighted clique graph was defined such that the weight of the maximum weight matching is equal to the weight of the perfect set defined by Algorithm 3. Thus, given a maximum weight matching, Algorithm 3 will construct a maximum perfect set:
PerfectSet
Input: A genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document} and a matching M.
add the adjacencies that corresponds to u and v to P
10
end
11
end
12
returnP
Lemma 3
Given a genome\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}, let M be a matching of\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document}and let\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P = \textsc{PerfectSet} (\tilde{G}, M)$$
\end{document}. If M is a maximum weighted matching then P must be a quasi maximum perfect set.
Proof
Assume towards contradiction that M is a maximum weight matching but P is not a quasi maximum perfect set. Then there must exist a perfect set P′ such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal W} (P) < {\cal W} (P^{\prime})$$
\end{document}. Let M′ be the matching that correspond to P′. Since the weight of the perfect set and its corresponding matching are the same, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal W} (M) < {\cal W} (M^{\prime})$$
\end{document}. But M is a maximum weight matching, a contradiction. ▪
While in many cases the maximum weight matching will correspond to a quasi maximum perfect set that also covers the genome, simply finding a maximum weight matching of the fully modified weighted clique graph does not guarantee that the perfect set will be covering. Fortunately, any matching can be easily modified by Algorithm 4 so that this is the case:
ExtendedMaximumWeightMatching
Input: A fully modified weighted clique graph \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document} of a genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Output: A maximum weight matching M of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document} where every non-null vertex in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document} is incident with an edge in M.
foreach non-null vertex v in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document}do
3
ifv is not incident with an edge in Mthen
4
let u be the null vertex adjacent to v
5
add {u, v} to M
6
end
7
end
8
returnM
Lemma 4
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \textsc{ExtendedMaximumWeightMatching} ({\cal CG} (\tilde{G}))$$
\end{document}, where\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}is an unordered genome, is a maximum weight matching such that\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P = \textsc{PerfectSet} (\tilde{G}, M)$$
\end{document}covers\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Proof
Since every non-null vertex corresponds to an extremity in a genome, the perfect set will only be covering if every non-null vertex is incident with an edge in the matching from which the perfect set was created.
Algorithm 4 first finds any maximum weight matching M′. If every non-null vertex is incident with an edge in M′ then the algorithm does not modify the matching and the result is a maximum weight matching whose corresponding perfect set covers \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Each non-null vertex has a null vertex to which only it is adjacent. For non-null vertices not incident in an edge in the maximum weight matching, Algorithm 4 adds the edge between this vertex and its null counterpart to the matching. Clearly, the resulting perfect set will cover \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. Thus, we must ensure that the resulting set of edges is still a matching of maximum weight.
The resulting set of edges must be a matching since the non-null vertex is not incident with any edges in the matching and the null vertex is only adjacent with the non-null vertex. It must be a maximum weight matching since all weights in a fully modified weighted clique graph are non-negative.
Therefore, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \textsc{ExtendedMaximumWeightMatching} ({\cal CG} (\tilde{G}))$$
\end{document} is a maximum weight matching such that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P = \textsc{PerfectSet} (\tilde{G}, M)$$
\end{document} covers \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}. ▪
7. Algorithm
From Lemmas 3 and 4, we can conclude the following theorem:
Theorem 1
Let\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \textsc{Extended Maximum Weight Matching} \ ({\cal CG} (\tilde{G}))$$
\end{document}and\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$P = \textsc{PerfectSet} (\tilde{G}, M)$$
\end{document}, where\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}is an unordered genome. P is a quasi maximum perfect and covering set of\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G}$$
\end{document}.
Algorithm 5 is the complete breakpoint aliquoting algorithm bringing together all the algorithms discussed in the previous sections. It follows from Lemma 2 and Theorem 1 that Algorithm 5 produces the optimal breakpoint aliquoting.
BreakpointAliquoting
Input: A genome G with all gene families of size p.
Output: A polyploid H with all gene families of size p.
Most of the algorithms discussed in this article run in linear time, however, the best known algorithm for computing the maximum weight matching runs in O(ev log v) time (Lovász and Plummer, 2009) where e is the number of edges and v is the number of vertices. If we have a genome with n gene families of size p, the fully modified weighted clique graph will have 4n vertices and (2p − 2)n edges. Thus, our algorithm runs in O(pn2 log n) time.
8. Example
Suppose the genome \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G = \{\{ \stackrel{\succ}{a_1}\}, \{ \vec{a_1}, \stackrel{\succ}{b_1}\}, \{ \vec{b_1}, \vec{c_1}\}, \{ \stackrel{\succ}{c_1}, \vec{a_2}\}, \{\stackrel{\succ}{a_2}, \vec{c_2}\}, \{\stackrel{\succ}{c_2},\vec{d_1}\}, \{\stackrel{\succ}{d_1}\}, \{\stackrel{\succ}{d_2}\},\{ \vec{d_2}, \stackrel{\succ}{c_3}\}, \{ \vec{c_3}, \stackrel{\succ}{a_3}\}, \{ \vec{a_3}, \stackrel{\succ} {b_2}\}, \{ \vec{b_2}\}, \{ \vec{b_3}\}, \{ \stackrel{\succ}{b_3}, \vec{d_3}\}, \{ \stackrel{\succ}{d_3}\} \}$$
\end{document} is given as input to Algorithm 5. All the gene families of G contain three genes, hence, we hope to reconstruct a hexaploid ancestor, an ancestor with three different types chromosomes for a total of six chromosomes, of G using the algorithm. Its corresponding unordered genome is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{G} = \{\{\stackrel{\succ}{a}\}, \{ \vec{a}, \stackrel{\succ}{b}\}, \{ \vec{b}, \vec{c}\}, \{ \stackrel{\succ}{c}, \vec{a}\}, \{\stackrel{\succ}{a}, \vec{c}\}, \{\stackrel{\succ}{c}, \vec{d}\}, \{ \stackrel{\succ}{d}\}, \{\stackrel{\succ}{d}\}, \{ \vec{d}, \stackrel{\succ}{c}\}, \{ \vec{c}, \stackrel{\succ}{a}\}, \{ \vec{a}, \stackrel{\succ}{b}\}, \{ \vec{b}\}, \{ \vec{b}\}, \{\stackrel{\succ}{b}, \vec{d}\}, \{\stackrel{\succ}{d}\}\}$$
\end{document} and its fully modified weighted clique graph is depicted in Figure 1.
From Figure 1, we can see that the maximum weight matching of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (\tilde{G})$$
\end{document} consists of the set of edges \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$M = \{ \{ \vec {a}, \stackrel{\succ}{b} \}, \{ \stackrel {\succ} {c}, \vec {d} \}, \{ {\rm null}, \stackrel{\succ} {d} \}, \{ \stackrel{\succ} {a}, \vec {c} \}, \{{\rm null}, \vec {b} \} \}.$$
\end{document} As is often the case, in this particular example, all the extremities are present so extending the matching with Algorithm 4 adds nothing to M.
Remark 1
As an interesting aside, we observe that at this point it is possible to calculate the distance between G and its most parsimonious polyploid ancestor. While the perfect set is not computed until the next step of the algorithm, the weight of the perfect set is known now. From Lemma 3, the sum of weights of the edges of\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal CG} (G)$$
\end{document}is equivalent to the weight of the perfect set. Since the weight of the perfect set is based on the breakpoint distance equation, we can insert the weight of the matching perfect set into the equation by subtracting it from\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal G} (G)$$
\end{document}. The result is the breakpoint distance between G and its most parsimonious polyploid ancestor.
We can calculate the final breakpoint distance given the weight of M. Since \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal W} (M) = 8.5 \ {\rm and} \ {\cal G} (G) = 12$$
\end{document}, the breakpoint distance between G and the hexaploid we will construct will be 3.5.
Finally, Algorithm 2 is applied to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}. \ \{\stackrel{\succ}{d} \} \ {\rm in} \ \tilde{H}$$
\end{document} has three ordered equivalents in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$G: \{ \stackrel{\succ} {d_1}\}, \{\stackrel{\succ}{d_2}\}, \{\stackrel{\succ} {d_3}\}$$
\end{document}. The rest of the adjacencies in \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\tilde{H}$$
\end{document} each have two ordered equivalents in G, for example, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\stackrel{\succ} {c}, \vec {d} \}$$
\end{document} is equivalent to \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\stackrel{\succ}{c_2}, \vec {d_1} \} \ {\rm and} \ \{ \vec {d_2}, \stackrel{\succ}{c_3} \}$$
\end{document}. Since all the gene families are of size three and for every adjacency two of the three ordered adjacencies have equivalents in G, it is trivial to calculate the third ordered adjacency, in the case of \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \stackrel{\succ}{c}, \vec {d} \}$$
\end{document} it is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{\stackrel{\succ}{c_1}, \vec {d_3} \}$$
\end{document}. However, even in more complex examples, the remaining adjacencies are just as easy to calculate as the pairing of the unused ordered extremities is arbitrary. The resulting ordered hexaploid is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$H = \{ \{ \vec {b_1} \} \{ \vec {a_1}, \stackrel{\succ}{b_1} \}, \{ \stackrel {\succ}{a_1}, \vec {c_1} \}, \{ \stackrel{\succ} {c_1}, \vec {d_3} \}, \{ \stackrel{\succ} {d_3} \}, \{ \vec {b_2} \}, \{ \vec {a_3}, \stackrel{\succ} {b_2} \}, \{ \vec {c_3}, \stackrel{\succ} {a_3} \}, \{ \vec {d_2}, \stackrel{\succ} {c_3} \} \{ \stackrel{\succ} {d_2} \}, \{ \vec {b_3} \}, \{ \vec {a_2}, \stackrel {\succ} {b_3} \}, \{ \stackrel {\succ} {a_2}, \vec {c_2} \}, \{ \stackrel{\succ} {c_2},\vec {d_1} \}, \{ \stackrel {\succ} {d_1}\} \}$$
\end{document}.
Since both H and G are ordered we can compare them. The only breakpoints are in H are \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\{ \vec {b_1} \}, \{ \stackrel{\succ} {a_1}, \vec {c_1} \}, \{ \stackrel{\succ} {c_1}, \vec {d_3} \} \ {\rm and} \ \{ \vec {a_2}, \stackrel{\succ}{b_3} \}$$
\end{document}; since \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal A} (G, H) = 6 \ {\rm and} \ {\cal T} (G, H) = 5 \ {\rm and} \ {\cal G} (G) = 12$$
\end{document}, from Equation 2, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} = 3.5$$
\end{document}. Observe that this matches perfectly with our earlier calculation.
9. Double Cut and Join Distance
Double cut and join distance and breakpoint distance are closely related. BP distance counts the number of differences between two genomes. For the most part, this is the same as DCJ distance; it generally takes one DCJ operation to correct one difference between two genomes. However, sometimes a DCJ operation corrects two differences instead of one, and the DCJ distance must take this into account; this is the only difference between the two distances and it is not very significant as this occurs infrequently with most data sets encountered in practice.
The double cut and join (DCJ) operation acts on two adjacencies or telomeres u and v of a genome in one of the following three ways:
• If both u = {w, x} and v = {y, z} are adjacencies, these are replaced by the two adjacencies {w, y} and {x, z} or by the two adjacencies {w, z} and {x, y}.
• If u = {w, x} is an adjacency and v = {y} is a telomere, these are replaced by {w, y} and {x} or by {x, y} and {w}.
• If both u = {w} and v = {x} are telomeres, these are replaced by {w, x}.
In addition, as an inverse of the last case, a single adjacency {w, x} can be replaced by two telomeres {w} and {x}.
DCJ distance is easily computed with the aid of the following graph:
An adjacency graph (depicted in Fig. 2) is a bipartite graph with two sets of vertices labeled with the elements of the two genomes that are to be compared respectively. There is an edge connecting two vertices for each extremity in common between their corresponding adjacencies.
An example of an adjacency graph.
Recall that to be compared using a distance algorithm like DCJ, both genomes must have the same genes, hence the same extremities, and, in the case of genomes with duplicated genes, must be ordered.
From the adjacency graph, the DCJ distance can be computed:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{\cal D} _ {DCJ} (G, H) = | {\cal G} (G) | - c - \frac{i} {2} \tag {3}
\end{align*}
\end{document}
where c is the number of cycles and i is the number of paths with an odd number of edges in the adjacency graph.
The following theorem gives the relationship between the DCJ distance and BP distance:
Theorem 2
Given two genomes G and H:\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}
{\cal D}_{DCJ} (G, H) \leq {\cal D}_{BP} (G, H) \leq 2 \cdot {\cal D}_{DCJ} (G, H) \tag{4}
\end{align*}
\end{document}
Proof
First, for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{DCJ} (G, H) \leq {\cal D}_{BP} (G, H)$$
\end{document}, observe that every adjacency shared between the two genomes causes a cycle of size 2 in the adjacency graph. It follows that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal A} (G, H)|$$
\end{document} is equal to the number of cycles of size 2 in the adjacency graph and, hence, less than or equal to the number of cycles in the adjacency graph. By similar reasoning, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal T} (G, H)|$$
\end{document} is equal to the number of paths of size 1 in the adjacency graph and, hence, less than or equal to the number of paths in the adjacency graph. It follows that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal A} (G, H)| + \frac{|{\cal T} (G, H)|}{2} \leq c + \frac{i} {2}$$
\end{document} where c is the number of cycles and i the number of odd paths in the adjacency graph. Since this factor decreases the distance, it follows that \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{DCJ} (G, H) \leq {\cal D}_{BP} (G, H)$$
\end{document}.
Second, for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H) \leq 2 \cdot {\cal D}_{DCJ} (G, H)$$
\end{document}, we will assume the most extreme case, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal A} (G, H)| + \frac{| {\cal T} (G, H)|} {2} = 0$$
\end{document} but the number of cycles and paths in the adjacency graph is otherwise maximal. This produces the worst possible BP distance, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal G} (G, H)|$$
\end{document}, but it is otherwise a maximal DCJ distance.
To determine the maximum number of cycles and paths in the adjacency graph, observe that every edge in the adjacency graph corresponds to one extremity shared between the genomes. Since \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$|{\cal A} (G, H)| + \frac{|{\cal T} (G, H)|} {2} = 0$$
\end{document} it follows that there are no cycles of size 2 and odd paths of size 1, so the smallest cycles and odd paths are of size 4 (since all cycles are even) and 3 respectively. Thus, the maximum number of cycles and odd paths is \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\frac{|{\cal G} (G, H)|} {2}$$
\end{document}. Thus, in the worst case scenario, BP distance is equal to twice the DCJ distance. ▪
Because BP distance can be equal to the DCJ distance, it is possible that the genome that results from Algorithm 5 could represent the optimal aliquoting for both BP and DCJ. Because \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H) \leq 2 \cdot {\cal D}_{DCJ} (G, H)$$
\end{document}, the distance between the original genome and its optimal BP aliquoting is no more than twice the distance between the original genome and its optimal DCJ distance. Thus, we can deduce the following:
Theorem 3
Algorithm 5 is a 2-approximation for the genome aliquoting problem using DCJ distance.
We can see that Theorem 3 holds true in the case of the example discussed in Section 7. Figure 3 depicts the adjacency graph between the genomes G and H from Section 7. As we can see, c = 7, i = 6 and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal G} (G) = 12$$
\end{document}; hence, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{DCJ} (G, H) = 2$$
\end{document}. While \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{BP} (G, H) = 3.5$$
\end{document}, close to the upper bound of twice \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal D}_{DCJ} (G, H)$$
\end{document}, the resulting double cut and join distance is very good and, in fact, is almost certainly optimal.
An adjacency graph between an order genome with all gene families of size three (top) and one possible hexaploid ancestor (bottom) where the distance between the two genomes is minimal.
10. Conclusion
Since the input to our algorithm is of size pn where p is the size of the gene families and n is the number of gene families, our algorithm runs in sub-cubic time. Thus, there exists a polynomial time algorithm that solves the genome aliquoting problem for BP distance. However, our algorithm is not without limitations.
The output of our algorithm consists of a mixture of linear and circular chromosomes with the only restriction being that adjacencies of the form {x, x} for some extremity x are not permitted. In many cases, it is desirable to restrict the output to something more similar to the input. For example, if the input consists of linear chromosomes then the output should consist of linear chromosomes. We believe it is possible to modify the output of our algorithm to provide the desired output in most cases without changing the distance. Some of the genome halving algorithms do precisely this (El-Mabrouk and Sankoff, 2003; Alekseyev and Pevzner, 2007). On the other hand, sometimes circular chromosomes with adjacencies of the form {x, x} are desirable in the output, in which case it might be possible, in some cases, to get a better result than produced by our algorithm.
It remains an open problem whether or not a polynomial time algorithm for the genome aliquoting problem with DCJ distance exists. However, as we have shown, a good approximation algorithm does exist. We remain optimistic that a polynomial time algorithm for DCJ distance can be found in the future.
Footnotes
Acknowledgments
Research was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada. David Sankoff holds the Canada Research Chair in Mathematical Genomics.
Disclosure Statement
No competing financial interests exist.
References
1.
AlekseyevM.A., PevznerP.A.2007. Whole genome duplications and contracted breakpoint graphs. SIAM J. Comput., 36:1748–1763.
2.
BergeronA., MixtackiJ., StoyeJ.2006. A unifying view of genome rearrangements. Lect. Notes Comput. Sci., 4175:163–173.
3.
El-MabroukN., SankoffD.2003. The reconstruction of doubled genomes. SIAM J. Comput., 32:754–792.
4.
FeijãoP., MeidanisJ.2009. SCJ: a novel rearrangement operation for which sorting, genome median and genome halving problems are easy. Lect. Notes Comput. Sci., 5724:85–96.
5.
LovászL., PlummerM.D.2009. Matching Theory. AMS Chelsea Publishing: Providence.