
Abstract
Abstract
We consider the following problem: given a set of gene family trees, spanning a given set of species, find a first speciation which splits these species into two subsets and minimizes the number of gene duplications that happened before this speciation. We call this problem the Minimum Duplication Bipartition Problem. Using a generalization of the Minimum Edge-Cut Problem, we propose a polynomial time 2-approximation algorithm for the Minimum Duplication Bipartition Problem. We apply this algorithm to the inference of species trees on synthetic datasets and on two datasets of eukaryotic species.
1. Introduction
Recently, Chauve and El-Mabrouk (2009) and Scornavacca et al. (2011) described a formal link between the Minimum Duplication Problem and a problem of supertrees (Bininda-Edmonds, 2004), where, given a set of uniquely leaf-labeled gene trees (there is at most one copy of each gene in each genome), the goal is to reconstruct a species tree that disagrees with the minimum number of gene trees (Byrka et al., 2010). This problem—a version of the MD Problem restricted to uniquely leaf-labeled trees—is NP-hard too, even in the simple case where each gene tree is a rooted triplet (Bryant, 1997). For supertree problems, greedy heuristics based on the principle of computing successive optimal speciations, modeled as edge-cuts in a graph whose vertices are the considered species, are now standard (Page, 2002; Semple and Steel, 2000). In such heuristics, each Minimum Edge-Cut splits the set of considered species into two subsets, corresponding to a speciation which results in two distinct lineages. Computing an optimal first speciation (a first speciation that disagrees with the least number of rooted triplets) is tractable, as the Minimum Edge-Cut problem is tractable. A complete species tree can then be obtained from a series of locally optimal speciations.
In the present work, we consider the Minimum Duplication Bipartition (MDB) Problem: given a set of gene trees (where a gene occurs any number of times), find a bipartition of the considered genes (corresponding to a speciation) that minimizes the number of duplications that happened before this speciation. A first motivation for this problem is that it leads, as for supertree problems, to natural greedy heuristics to reconstruct a species tree from a set of gene trees. Our work is also motivated by understanding the combinatorial properties of speciations with respect to gene duplications. Our main result is a polynomial time 2-approximation algorithm for the Minimum Duplication Bipartition Problem that generalizes the Minimum Edge-Cut approach used in supertrees. Although our focus here is primarily theoretical, we also provide experimental results, on small simulated datasets, and two real eukaryotic large-scale datasets.
We first define, in Section 2, gene trees, species trees, duplications, and the optimization problems considered in this article. In Section 3, we show how the Minimum Duplication Bipartition Problem can be described as a variant of the classical Minimum Edge-Cut Problem. Then we use this description to reduce the Minimum Duplication Bipartition Problem to a Set Function Minimization Problem. We finally describe a 2-approximation algorithm through a Minimum Hypergraph Cut Problem. In Section 4, we describe results of experiments on simulated and real eukaryotic data, illustrating our algorithm in a realistic phylogenomics context.
2. Preliminaries: Objects, Problems, Background
Consider,
Gene and species trees, bipartitions
A species tree on
Given a tree T whose leaves are labeled by integers from
A bipartition B on a set S is a partition of S into two subsets S1 and S2. We represent a bipartition by a, possibly non-binary, species tree on S containing exactly three internal vertices—the root v and its two children vℓ and vr—such that L(vℓ) ∩ L(vr) = ∅, L(vℓ) ∪ L(vr) = S, L(vℓ) = S1 and L(vr) = S2.
Reconciliation between Gene Trees and Species Trees
The Lowest Common Ancestor Mapping (LCA mapping) is central in the problem of reconciling a gene tree and a species tree. Given a gene tree T and a species tree S on
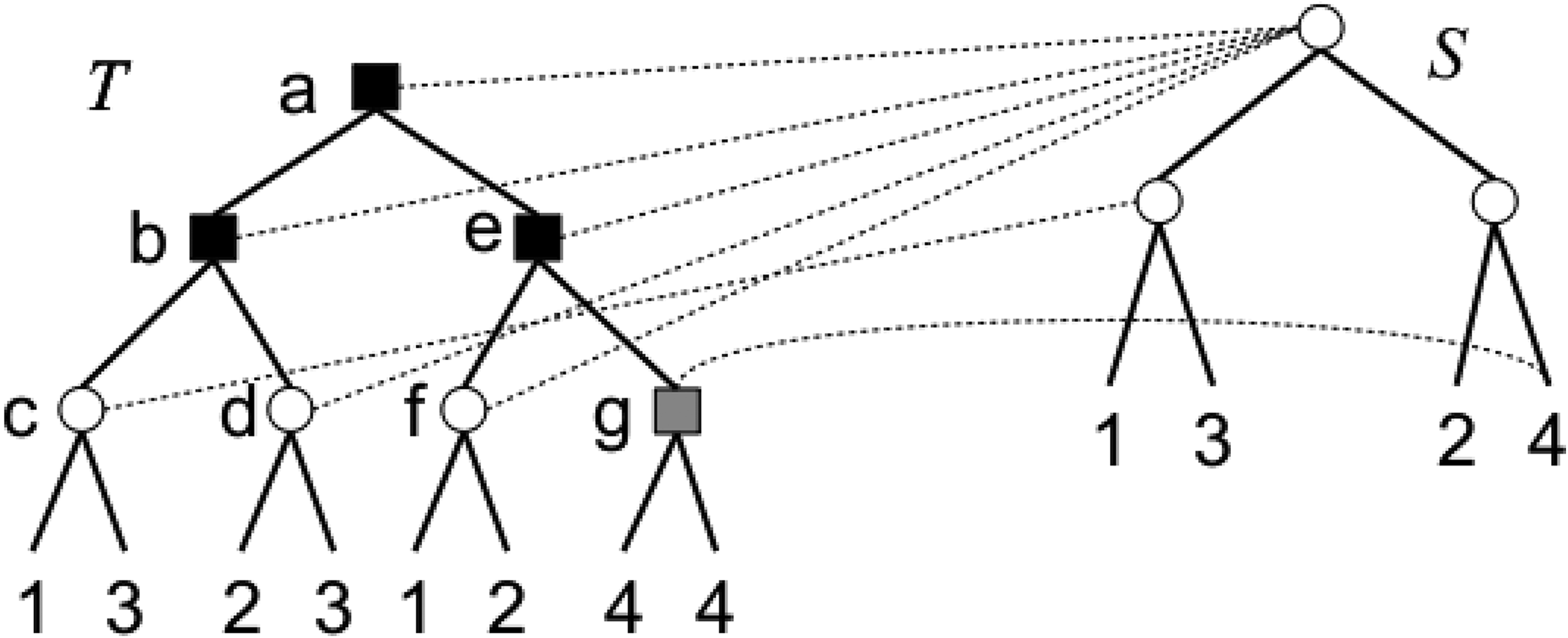
A gene tree T and a species tree S on a set of genome
If L(xℓ) ∩ L(xr) ≠ ∅, then x is a duplication vertex with respect to any species tree S on
Inferring parsimonious species trees and speciations
It is well known that d(F, S) is the minimum number of gene duplication events required in any evolutionary scenario that resulted in F (Chauve and El-Mabrouk, 2009; Górecki and Tiuryn, 2006), which leads to the following optimization problem called the Minimum Duplication Problem (MD Problem): given a gene tree forest F, find a species tree S such that d(F,S) is minimum.
The MD Problem is NP-hard (Ma et al., 2000), even in the case where every gene tree is a uniquely leaf-labeled and rooted triplet (Bryant, 1997), in which case it is in fact equivalent to a supertree problem called the Minimum Rooted Triplet Inconsistency (MRTI) Problem. This link with supertrees is important as recent hardness results on the MRTI Problem imply that
Hence, for solving the MD Problem, one has to rely on exponential time algorithms (Hallett and Lagergren, 2000) or local-search methods with no optimality guarantee (Bansal et al., 2007; Wehe et al., 2008).
In Chauve and El-Mabrouk (2009), it was shown that the MD Problem is in fact a variant of a supertree problem (see also Scornavacca et al. [2011], which explores the link between gene duplications and supertrees). Greedy heuristics for hard supertree problems based on computing successive speciations events have proved to be effective (Semple and Steel, 2000), and in Chauve and El-Mabrouk (2009), an application of such a heuristic showed promising results on synthetic data. This motivates the introduction of the main problem we study in this article:
Before discussing previous works, we state an obvious, but useful, property related to duplication vertices of a forest of gene trees F on
Property 1
Let x be a vertex of F. Given a bipartition B on
Previous work
As far as we know, the MDB problem was introduced in Stege (1999), where an exponential time algorithm was proposed. Unlike the MD Problem, it is obviously FPT, as there are
Related optimization problems
Given a connected graph G = (V, E), an edge-cut of G is an edge set E′ ⊆ E whose removal disconnects the graph G. A bipartition B with root v, on the set of vertices of G, induces a unique edge-cut of G, denoted by EG(B), composed of the edges
The Minimum Edge-Cut (MEC) Problem asks for a bipartition on the vertices of G inducing an edge-cut of G of minimum cardinality. If the edges of G are labeled on a given set Σ of labels, given an edge-cut E′ of G, the label-set of E′ denoted by label(E′) is the subset of Σ composed of the labels of the edges in E′. The following cut problem is a natural generalization of the MEC Problem and is essential in our algorithm:
A set function is a function
Remark 1
The Minimum Edge-Cut problem is a special case of the SFM Problem: given a graph H = (V, E), if we denote by g the cut-set function defined from 2
V
to
A hypergraph is a pair (V, E) where V is a set of vertices and E is a set of non-empty subsets of V called hyperedges. Given a hypergraph G = (V, E), a bipartition B on V with root v induces a hyperedge-cut of G defined by the following subset of E:
3. A 2-approximation algorithm for the MDB Problem
We first describe how the MDB problem can naturally be translated into an edge-cut problem; however the tractability of this edge-cut problem is unknown, as it cannot be reduced to an SFM problem. Next, we describe our main result, that is an efficient 2-approximation algorithm for the MDB problem.
From now on, given a gene tree forest, we label arbitrarily its internal vertices with a set Σ of labels in such a way that no two internal vertices have the same label.
3.1. A set function minimization problem
Given a gene tree forest F, we define the edge-labeled graph H(F) = (V, E) associated to F as follows (Fig. 2): V = L(F) and there is an edge labeled with
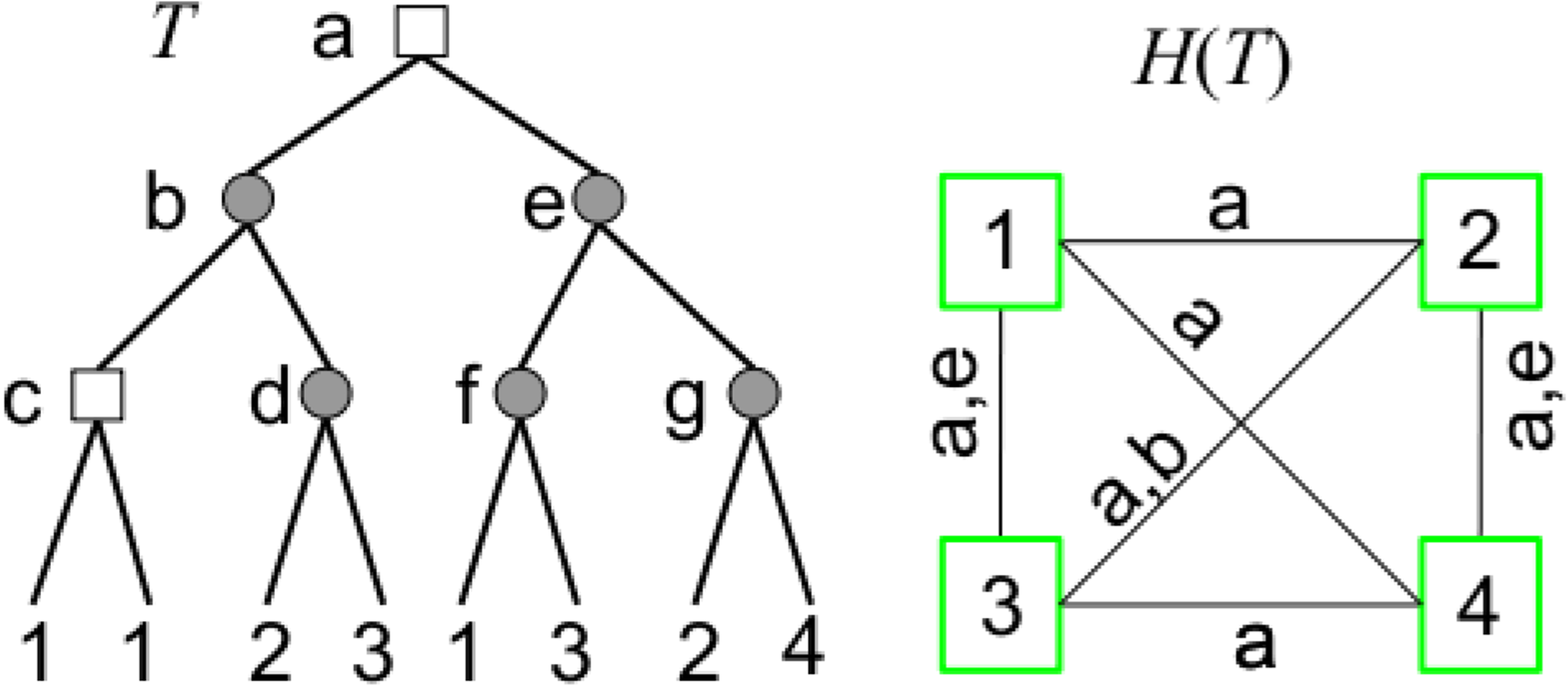
A gene tree T on the set of genomes
Lemma 1
Let F be a gene tree forest on
Proof
If a is the label of a pre-duplication x in F with respect to the bipartition B with root v, then, from Property 1, there is a pair
Remark 2
If a forest F of gene trees contains uniquely leaf-labeled triplets, then each label a appears on a single edge, and the MDB problem is equivalent to the Minimum Edge-Cut problem on H(F), and then tractable.
The MLEC Problem (and then the MDB Problem) can be reduced to a Set Function Minimization Problem as follows: given an edge-labeled graph G = (V, E), we define the cut-set function
Modeling the MDB problem as an edge-cut problem naturally leads to ask if this edge-cut problem is an instance of the SFM problem, in which case it would be tractable. As noted in Remark 1, the MLEC Problem is NP-hard. However, it is not difficult to see that the graphs used in Jha et al. (2002) to prove the hardness of the Minimum label Cut Problem can not be obtained from gene tree forests. Unfortunately, the cut-set function fG associated to an edge-labeled graph G is not always a submodular function, as shown below.
Property 2
There exists a gene tree forest F such that the cut-set function fH, where H = H(F), is not a submodular function.
For example (Fig. 3), consider a single gene tree T with four leaves {1, 2, 3, 4} and three internal vertices a, b and c whose sets of children are respectively {b, c}, {1, 3} and {2, 4}. The edge-labeled graph H(T) associated to T has four vertices {1, 2, 3, 4} and only two edges (1, 3) and (2, 4) labeled with a. If we consider the subsets A = {1, 4} and B = {2, 4} of the set of {1, 2, 3, 4}, we see that fH(T)(A) = 1, fH(T)(B) = 0, fH(T)(A ∪ B) = 1 and fH(T)(A ∩ B) = 1. Then, fH(T)(A) + fH(T)(B) = 1 < fH(T)(A ∪ B) + fH(T)(A ∩ B) = 2, and fH(T) is not a submodular function.
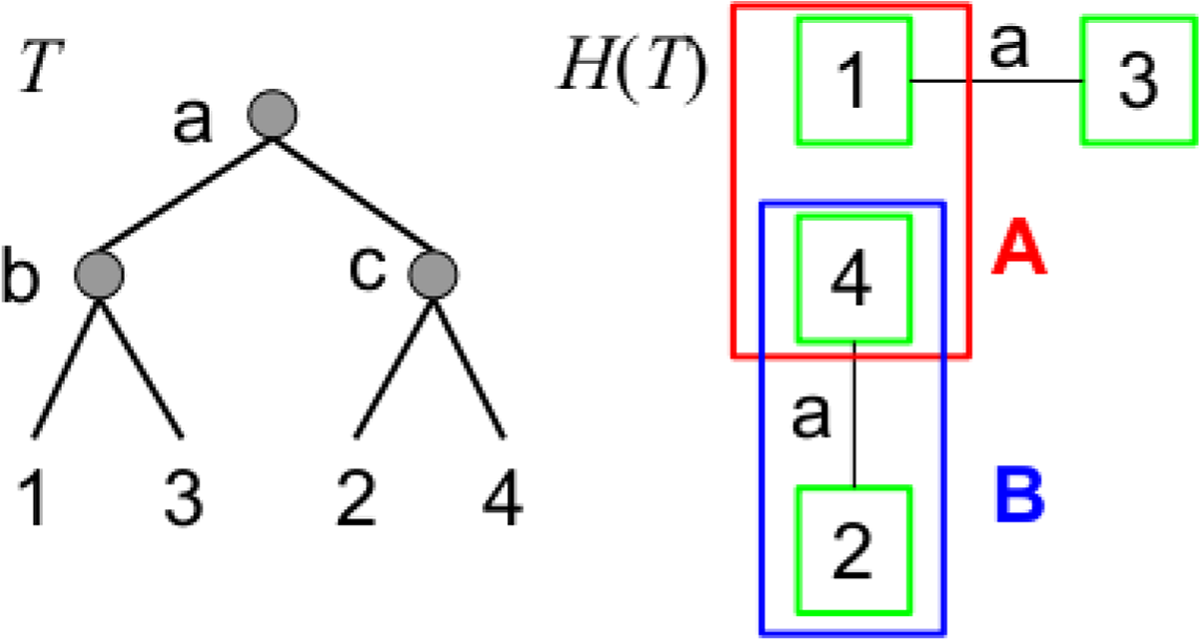
Illustration of Property 2: a gene tree T (left) and the corresponding graph H(T) (right), and two vertex sets A and B that contradict the submodularity of the cut-set function fH.
We described in Chauve and Ouangraoua (2009) and Ouangraoua et al. (2010) the necessary conditions for the cut-set function fH to be non-submodular. Based on these conditions, we were able to show that the graph H(F) can be augmented into a graph I(F) in such a way that the (labeled) cut-set function fI is submodular. This result, aside from its theoretical interest, lead to a 2-approximation algorithm for the MDB Problem, based on solving a SFM Problem on the graph I(F). In Section 3.2 below, we describe another 2-approximation through a direct link with the Minimum Hypergraph Cut problem, whose exposition is simpler.
3.2. An approximation through the Minimum Hypergraph Cut problem
Given a gene tree forest F, we define the hypergraph J(F) = (V, E) associated to F as follows (Fig. 4): V = L(F) and, for each apparent duplication x in F, L(x) is a hyperedge of J(F) and, for each non-apparent duplication y in F, L(yℓ) and L(yr) are two hyperedges of J(F).
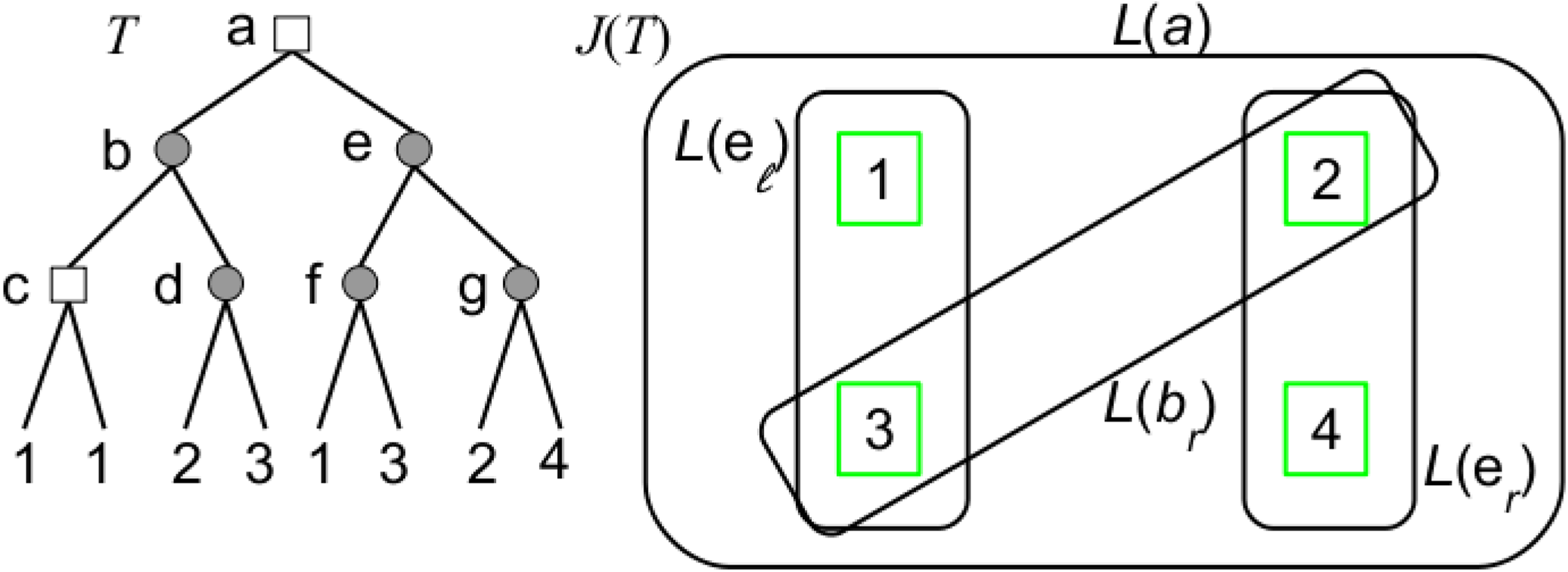
A gene tree T on the set of genomes
Theorem 1
Let F be a gene tree forest with n vertices, on a set
Proof
For any bipartition B on L(F), the cardinality of label(EH(B)) (resp. EJ(B)) is denoted by dH(B) (resp. dJ(B)).
We first prove that, for any bipartition B on L(F) with root v, dH(B) ≤ dJ(B) ≤ 2*dH(B). It is relatively straightforward. A label
Now, let B′ be a bipartition on L(F) inducing an optimal labeled edge-cut of H(F) (i.e., dH(B′) is minimum). For any bipartition B on L(F), if dH(B) > 2 * dH(B′) then B cannot induce an optimal hyperedge-cut of J(F): indeed, if dH(B) > 2 * dH(B′), as dH(B) ≤ dJ(B) and dJ(B′) ≤ 2 * dH(B′), we have dJ(B) ≥ dH(B) > 2 * dH(B′) ≥ dJ(B′). Hence, for any bipartition B on L(F) that induces an optimal hyperedge-cut for J(F), we have dH(B) ≤ 2 * dH(B′). This completes the proof that computing an optimal labeled edge-cut for J(F) achieves a ratio 2 approximation for the MDB Problem.
The time complexity stated in Theorem 1 follows from the algorithm described in Mak, (2005) solving the MHC Problem on a hypergraph G in time O(kn) where k (resp. n) is the number of vertices (resp. hyperedges) of G. ▪
We showed in Ouangraoua et al. (2010) that the approximation ratio of 2 is tight. The approximation algorithm also has the following straightforward algorithmic property.
Property 3
If there exists at least one parsimonious first speciation B′ such that all the corresponding pre-duplications are apparent duplications in F, then solving the MHC Problem on J(F) gives a parsimonious bipartition.
To prove Property 3, we only need to note that if a first speciation B′ induces only pre-duplications which are apparent, then B′ defines an optimal cut for both H(F) and J(F). Note however that this does not imply that the cut-set function for H(F) is submodular.
4. Experimental Results
We performed four different experiments.1 First, on several datasets of simulated gene families on 12 species (genomes) we studied the ability of the greedy approach—that infers a species tree by computing successive parsimonious speciations—to recover the exact species tree, using an exhaustive exploration of all possible speciations at each step (which was possible due to the fact we considered only 12 species). Second, on the same simulated datasets, we replaced the exhaustive exploration of all possible speciations at each step by our 2-approximation algorithm for computing a parsimonious speciation. Third, we applied the approximation algorithm on a real dataset of 6808 gene families from 23 fungal genomes. Last, we performed the same experiment on a real dataset of 18584 gene families from 50 eukaryotic genomes.
4.1. Experiment on simulated datasets
Datasets
We analyzed the four synthetic datasets that were studied in Chauve and El-Mabrouk (2009); each dataset contains 100 gene trees. Each gene tree was generated from a single ancestral gene with duplication (birth) and loss (death) rates computed using the software CAFE (Bie et al., 2006) from real Drosophilia gene families (Hahn et al., 2007). The duplication/loss rates range from 0.02 event/million years to 0.2 event/million years, and the number of duplication ranges from roughly 1000 (with rate 0.02) to roughly 6000 (see Table 1 in Chauve and El-Mabrouk [2009]).
ni, number of duplications preceding the ith speciation; n, total number of pre-duplications associated to each binary configuration.
The cases where our algorithm recovers the most parsimonious configurations (i.e. inducing a minimum global number of duplications) are marked with**.
To balance the fact that each gene tree originates from a single ancestral gene (and then has no duplication before the first speciation), and to consider datasets including gene families generated with different duplication/loss rates, we created duplications that happened before the first speciation by clustering the 400 gene trees of the four datasets into 100 clusters of random size, and for every such cluster of a given size, say k, we generated a random binary tree with k leaves and replaced each leaf of this tree by a gene tree of the cluster, which amounts to creating around 4 duplications that precede the first speciation. We repeated this experiment ten times, generating ten different datasets.
Results
On each of the simulated datasets, we first observed that the greedy heuristic that computes successive parsimonious speciations using an exhaustive exploration leads to the exact species tree (Fig. 5), i.e. the one that had been used to generate the synthetic gene trees. Despite the relatively modest size of our synthetic datasets (12 species), it illustrates the potential of this greedy heuristic in a phylogenomics context, especially as the heuristic described in Chauve and El-Mabrouk (2009) inferred a slightly incorrect species tree for the two datasets with the highest duplication/loss rates. Moreover, we observed that our approximation algorithm provided the exact species tree every time. We believe this result can be explained by the fact that most duplications that occurred during the generation of the gene trees are apparent duplications (Fig. 5).
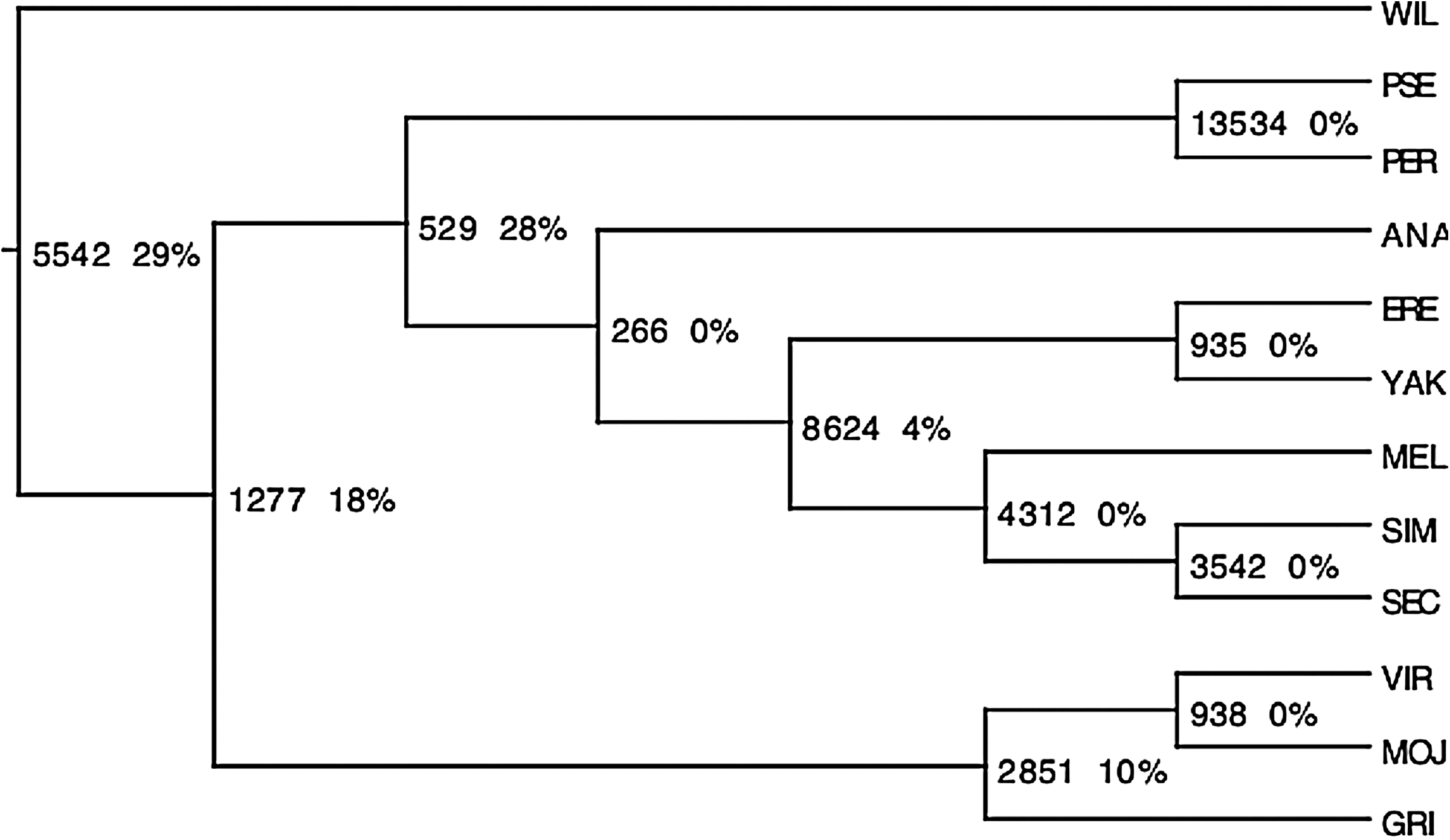
The species tree computed for the fourteen synthetic datasets on 12 species. Each speciation (vertex) of the tree is labeled with the total number of duplications preceding the speciation in all datasets, and the proportion of non-apparent duplications.
4.2. Experiment on a fungal dataset
Datasets
We uploaded from the Fungal Orthogroup Repository2 the 6808 fungal gene trees containing genes belonging to at least three different species, among 23 ascomycete fungal species.
Results
Due to the large number of species in the real data set—6808 fungal gene trees from 23 species—we applied only our approximation algorithm. We observed that the inferred species tree is the one widely accepted in yeasts phylogenomics3 (Fig. 6).
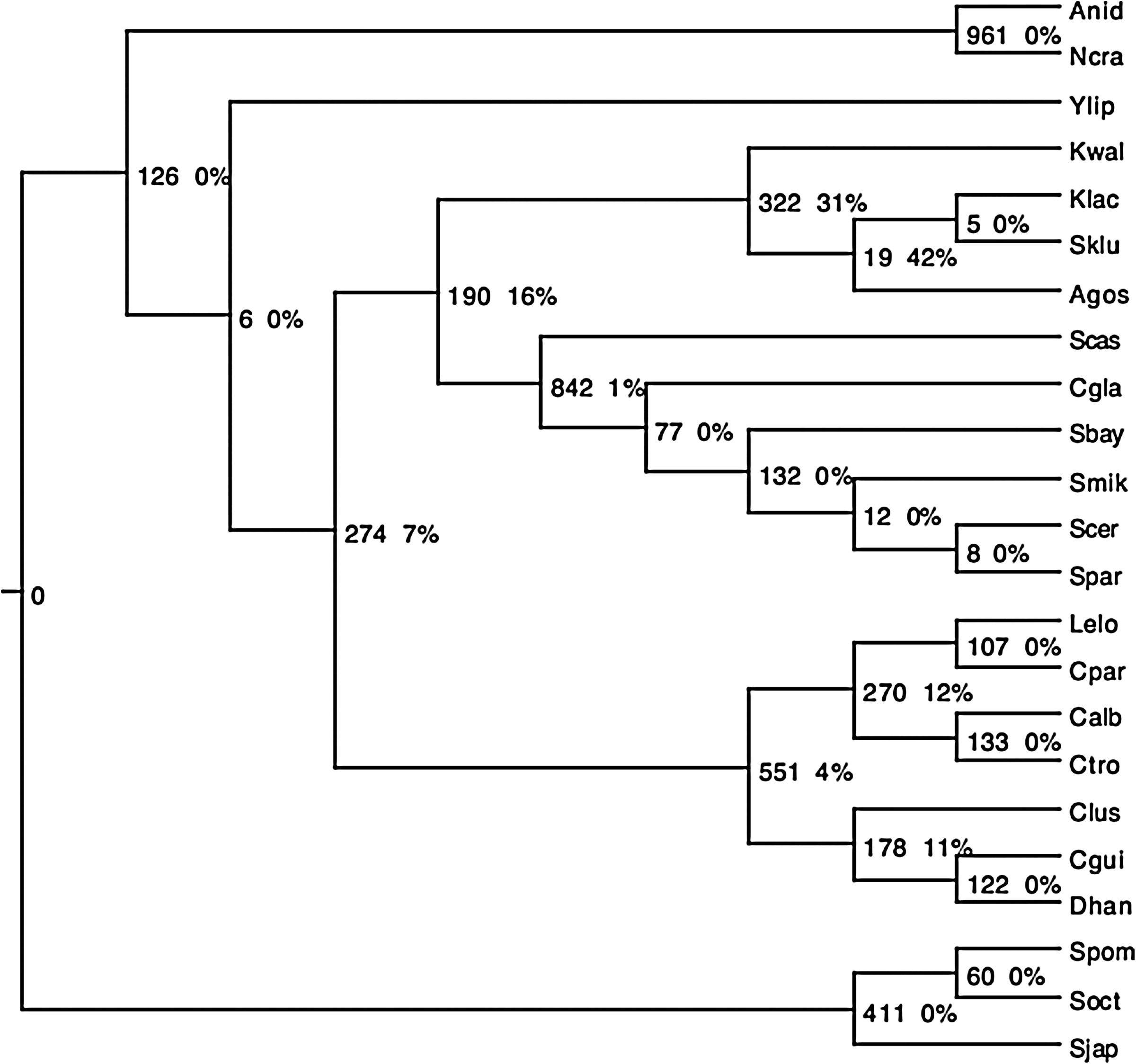
The species tree computed for the fungal datasets on 23 species. Each speciation of the tree is labeled with the number of duplications preceding the speciation (pre-duplications), and the proportion of non-apparent duplications. Species: A. nidulans (Anid), N. crassa (Ncra), Y. lipolytica (Ylip), K. waltii (Kwalt), K. lactis (Klac), S. kluyveri (Sklu), A. gossypii (Agos), S. castellii (Scas), C. glabrata (Cgla), S. bayanus (Sbay), S. mikatae (Smik), S. cerevisiae (Scer), S. paradoxus (Spar), L. elongosporus (Lelo), C. parapsilosis (Cpar), C. albicans (Calb), C. tropicalis (Ctro), C. lusitaniae (Clus), C. guilliermondii (Cgui), D. hansenii (Dhan), S. pombe (Spom), S. octosporus (Soct), S. japonicus (Sjap).
We also noticed that a significant number of speciations corresponded to apparent duplications only, while some of them are associated with a large number of duplications. The apparent duplications correspond to the speciation vertices labeled with 0% non-apparent pre-duplications in Figure 6. Such speciations are then parsimonious (see the discussion at the end of the previous section). Moreover, aside from one branch (leading to the group containing Kwal, Agos, Klac, Sklu) of the species tree, where 103 pre-duplications out of 322 (31% of the pre-duplications) are non-apparent, all other branches are associated with very few non-apparent pre-duplications, which suggests that they might be parsimonious. Providing the gene trees we analyzed are correct, this clearly suggests that traces of most duplications that occurred during yeast evolution are still visible today.
4.3. Experiment on a large-scale eukaryotic dataset
Datasets
We uploaded from the release 58 of the Ensembl database the 18584 eukaryotic gene trees (Vilella et al., 2009) containing genes belonging to 50 eukaryotic species, and obtained from CDS backtranslated protein-based multiple alignments (Vilella et al., 2009). The reference phylogeny of the 50 species is represented in Figure 7.
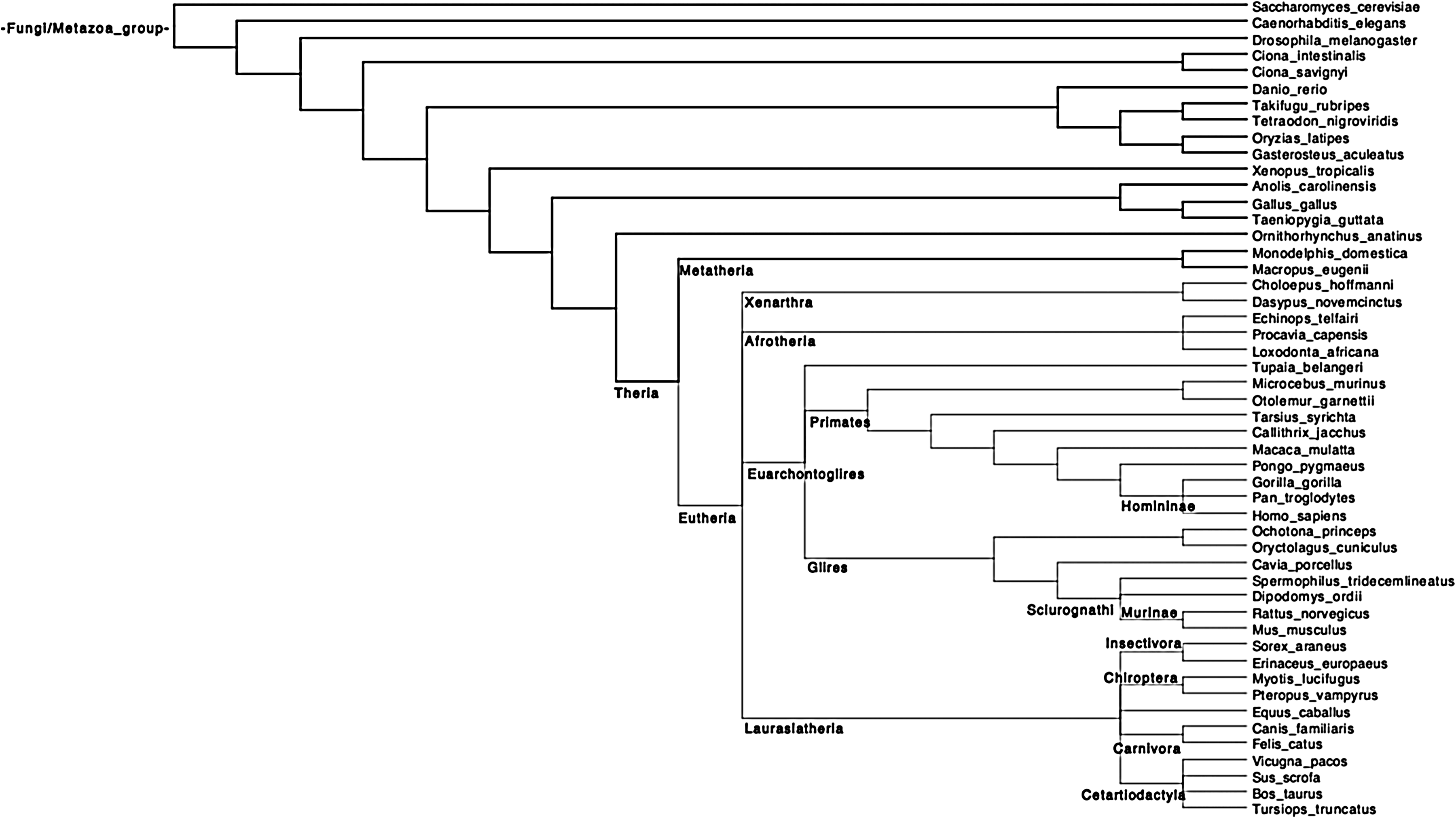
The 50 considered eukaryotic species. Tree obtained from the NCBI taxonomy at www.ncbi.nlm.nih.gov.
Results
We applied the same method as for the previous dataset, and obtained a species tree (Fig. 8) that is close to the one widely accepted in fungi/metazoa phylogenomics (Fig. 7). Among the 38 reference clades (i.e. set of species descending from the same internal vertex present in the reference tree), the inferred species tree recovers 35 clades, and does not recover only 3 clades inside the eutherian group (placental mammals): Xenarthra, Afrotheria, and Insectivora. However, the relative position of the species of these 3 clades compared to the others species remains congruent with recent studies on the phylogeny of placental mammals (Kjer and Honeycutt, 2007; Murphy et al., 2007; Nikolaev et al., 2008; Prasad et al., 2008).
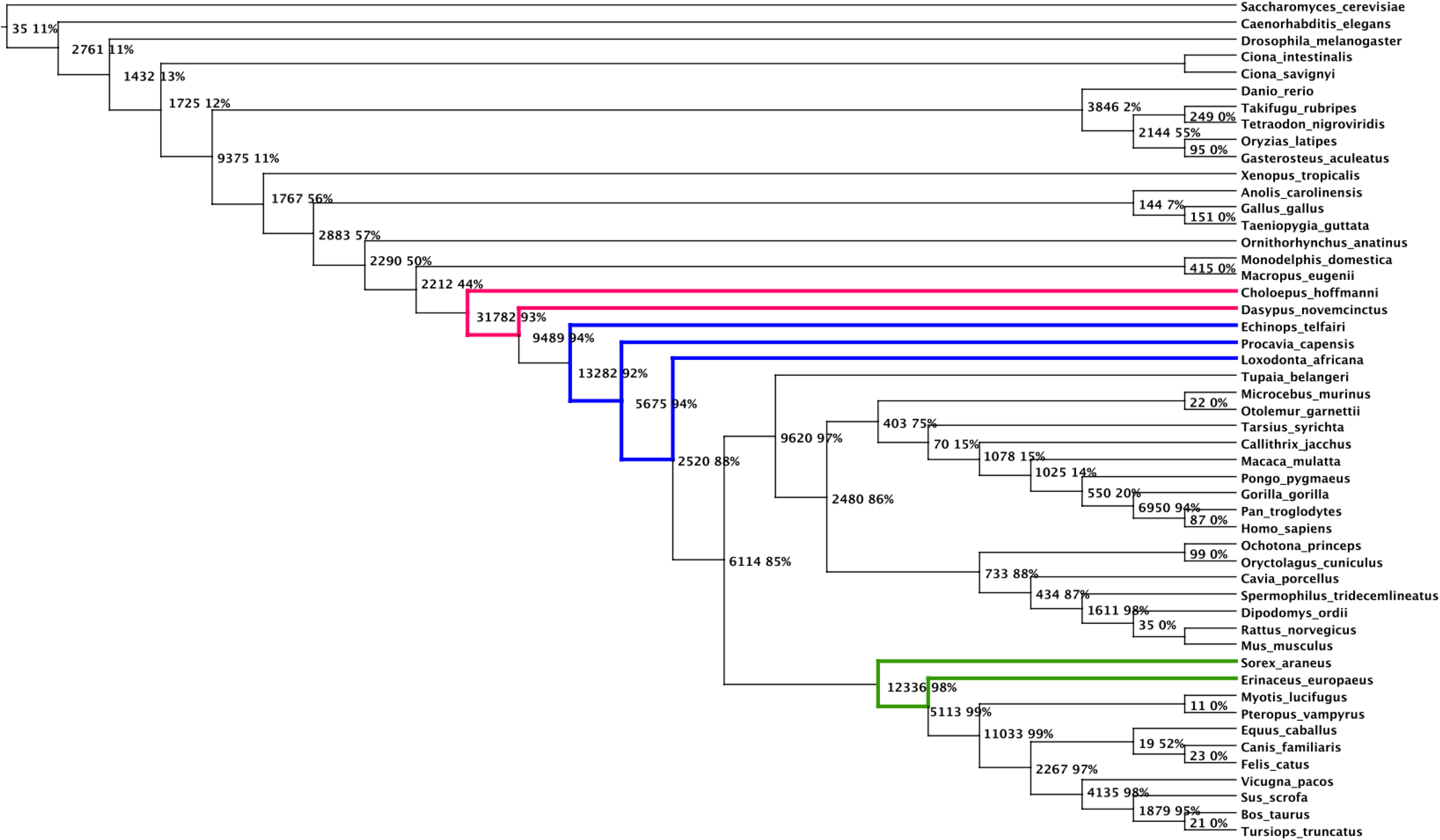
The species tree computed for the eukaryotic datasets on 50 species. Each speciation of the tree is labeled with the number of duplications preceding the speciation, and the ratio of non-apparent duplications. The clades that are missing regarding the NCBI reference tree are represented in red (Xenarthra), blue (Afrotheria), and green (Insectivora) colors.
First, we noticed that the two whole-genome duplications that occurred on the branch leading to the fish (Danio rerio, … , Gasterosteus aculeatus), and on the branch leading to vertebrates (Danio rerio, … , Tursiops truncatus), both left traces that are still visible today: a high number of pre-duplications (3846, 9375) with a low proportion of non-apparent duplications (2%, 11%).
From a structural point of view, we notice that the 3 speciations which are responsible for missing the monophyletic clades Xenarthra, Afrotheria, and Insectivora are characterized by very high numbers of pre-duplications (31782, 13282, 12336), coupled with high proportions of non-apparent duplications (93%, 92%, 98%). Moreover, all the speciations obtained for the unresolved nodes (i.e. nodes having more than two descending branches) of the reference tree (Eutheria, Euarchontoglires, Homininae, Laurasiatheria, and Cetartiodactyla) alse have high numbers of pre-duplications (more than 5000), coupled again with high proportions of non-apparent duplications (more than 90%).
In Table 1, we summarize the numbers of duplications associated with all the possible binary configurations for the unresolved nodes of the reference tree. We can observe that our algorithm misses the most parsimonious configuration for the roots of 4 clades: Eutheria, Afrotheria, Euarchontoglires, and Laurasiatheria. This is caused by the choice of a first speciation that has the lowest local pre-duplication cost, but leads to a non globally optimal configuration. These observations clearly suggests that considering a parsimony criterion in terms of gene duplications might not always be helpful to resolve difficult speciations. One explanation is that issues in reconstructing accurate and consistent phylogenetic trees might also impact the gene duplications signal, that is detected by reconciling gene trees and species trees. In these cases, the consideration of parsimonious k-furcations (where a speciation is the particular case where k = 2) instead of parsimonious speciations might help resolving the difficult phylogenies.
5. Conclusion
We showed that computing a parsimonious first speciation in the gene duplication model can be approximated in polynomial time with a ratio of 2. As far as we know this is the first time a constant approximation algorithm has been proposed in relation to the problem of inferring species trees using gene duplications. This result was obtained by describing the problem in terms of edge-cuts in particular graphs, which can be computed in polynomial time. This algorithm is also a natural generalization of the classical minimum edge-cut algorithm that is used in supertree consistency problems, which is highlighted by its link with the Submodular Function Minimization Problem.
From a theoretical point of view, the hardness of the Minimum Duplication Bipartition Problem is still an open problem, but we conjecture the problem is NP-complete. It is interesting to note that, as for the Gene Duplication Problem, when there is a parsimonious first speciation whose pre-duplications are all apparent duplications, it can be detected in polynomial time. Also when F contains only uniquely leaf-labeled rooted triplets, the graph H(F) does not need to be augmented as every label appears only once, and computing a parsimonious first speciation can be done by computing a minimum edge-cut in H(F). However, as we showed in the proof of Property 1, this tractability property no longer holds when quadruplets whose root is a non-apparent duplication are considered instead of triplets, as the cut-set function is no longer submodular. The role of non-apparent duplications, especially with respect to the non-submodularity of the cut-set function of H(F), seems to be fundamental to the hardness of the problem, in particular to the understanding of which families of gene tree forests are tractable or fixed-parameter tractable.
Our preliminary experiments showed that both the approach of inferring a species tree by computing successive parsimonious speciations and our approximation algorithm for computing such speciations are promising. However, we can notice that, on a large-scale eukaryotic dataset, some speciations, that were already considered as difficult to resolve by traditional phylogenetic methods, are associated with an unrealisticly large number of duplications and, more importantly, a large number of non-apparent pre-duplications. We investigated alternative hypothesis for such speciations, that resulted in comparable results. This suggests a possible way to characterize such difficult speciations, that are not supported by a strong signal in gene trees.
Footnotes
Acknowledgments
We thank Tamon Stephen, Mukul Bansal, Sylvain Guillemot, and Samuel Blanquart for useful discussions. Work was supported by an NSERC Discovery grant to C.C. and a fellowship from the ANR (project ANR-06-BLAN-0045) for A.O.
Disclosure Statement
No competing financial interests exist.