
Abstract
Abstract
1. Introduction
There are various reasons for the different types of noise in vivo. Many vital reactions happen between extracellular ligands, such as signal molecules, hormones, and receptors on the membrane of the cell, and between intracellular molecules and receptors on the membrane of organelles within the cell. Additionally, nutrients have to enter the cell by passing through the membrane and then interact with structural proteins and lipids. In other words, the mixing of molecules often can be obstructed.
Moreover, molecular reactions greatly depend on the availability of reaction volume. If a molecule, which participates in the reaction, is much smaller than the background molecules, then they have no, or only weak, influence on the reaction rate as the molecule can freely diffuse between them. On the other hand, the diffusion of molecules of similar size would be greatly hindered. This effect is called the volume exclusion effect (Schnell and Turner, 2004). In other words, a small molecule can easily occupy the volume in between obstacles, while the volume a molecule can occupy, whose size is of order of the obstacles, is greatly limited.
The success of systems biology depends on the ability to use computational models to unravel underlying biological mechanisms. These models often correspond to biological hypotheses, for example, about the underlying reaction network structure or the type and strength of noise. It is well known, however, that models of comparable complexity corresponding to different pathway connectivities may fit experimental data equally well (Roberts et al., 2009). Model-testing approaches that invalidate models can be used to reduce the number of possible models.
One shortcoming of current experimental design approaches for discriminating between rival mathematical models representing a biological system is that they often use a linearized version of models or cannot deal well with experimental noise and parameter uncertainties (August and Busch, 2011). Despite the fact that the cellular and intracellular environments are noisy, in silico model analysis for model-testing is mostly based on underlying models that are deterministic (see Mélykúti et al., 2010, and references therein). For these reasons, in this article we develop a novel model-testing approach based on “listening to the noise” (Munsky et al., 2009). We use the results of Øksendal (2003) and Kushner (1967) to present a state-of-the-art system analysis framework.
The structure of this article is as follows: In Section 2, we present different approaches to modeling and model-testing. We also show how the tests can be implemented computationally using new, state-of-the-art tools. For clarity, we describe these tools first, in Section 2.1. In Section 2.2, we briefly describe the deterministic modeling approach to biological systems, which is typically used in chemical reaction network theory (Feinberg, 1987), and a computational approach to test its stability.
However, because assuming deterministic dynamics is not always suitable for analyzing biological systems as argued previously, in Section 2.3 we introduce the notion of stochastic differential equations used for mathematical modeling of noisy systems. We present established results on estimating the exit probability of the solution trajectory of a system from a particular set in the phase space. We then provide a novel algorithm for model-testing using these results. Section 3 provides examples validating our approach to model-testing and its application to the Hog1 signaling pathway, allowing for the analysis of noise. We conclude the article in Section 4.
2. Methods
2.1. Semidefinite programming and the sum of squares decomposition
In this section, we provide some mathematical background on the computational tools we use for model-testing. These tools can also be used to computationally implement and solve other problems that arise in the field of model-analysis in biology (El-Samad et al., 2006b; August and Papachristodoulou, 2009). The main computational tool is semidefinite programming (Vandenberghe and Boyd, 1996), which can be solved efficiently using interior-point methods (Boyd and Vandenberghe, 2004). In semidefinite programming, we replace the nonnegative orthant constraint of linear programming by the cone of positive semidefinite matrices and pose the following minimization problem:
Here,
2.1.1. Sum of squares decomposition
For problem data that consists of polynomials of any degree, the requirement of positivity can be relaxed to the condition that the polynomial function is a sum of squares (SOS). On one hand, this is only a sufficient condition for positivity and can at times be quite conservative; in other words, a function can be positive without being an SOS. On the other hand, testing positivity of a polynomial can be NP-hard (Murty and Kabadi, 1987).
Consider the real-valued polynomial function F(
The length of vector
In this article, to solve SOS programs, we use SOSTOOLS (Prajna et al., 2002), a free, third-party MATLAB toolbox that relies on the solver SeDuMi (Sturm, 1999).
Note that the computational time necessary to solve sum of squares decomposition problems scales badly with the size of the problem (Table 1), which makes the application of our method to nonlinear models with more than five to six species prohibitive when using current solvers in MATLAB. Nonetheless, many models in biology do not consider more than six species.
The dimension of matrix
2.2. Deterministic modeling approach
Below, we briefly demonstrate the use of SOSTOOLS for checking system stability of a deterministic dynamical system used to represent a biological system. Consider the following set of ordinary differential equations representing a dynamical system with state vector
where function
However, as mentioned in the introduction, for modeling certain biological systems, a stochastic approach is more realistic. In the following section, we introduce such an approach and provide novel means using SOSTOOLS to distinguish between different models affected by noise by comparing them with measurement data.
2.3. Stochastic modeling approach
Consider the following stochastic differential equation representing a dynamical system with random state vector
where
2.3.1. Exiting a set
For
then the expected value of V(
where
Let
2.3.2. Using set exit for model-testing
We consider a stopped process (Prajna et al., 2004) and a model of the form given by Eq. (5) for a system under observation. For initial condition
then the model predicts an upper bound on the probability that is lower than Pm, which is the measured one, and, thus, invalidates the model.
Generally, one must be very lucky if setting V(
Theorem 1.
Consider a model of the form given by Eq. (5) for a system under observation, an initial condition
Proof.
Note that
Eq. (12) guarantees that if f (
Eq. (13) guarantees that if
Eq. (14) guarantees that if ∥
Hence, if the problem given by Eqs (9)–(14) has a solution then
but since
this invalidates the model. ▪
3. Results
3.1. Two structurally different two-dimensional systems
3.1.1. Exit from a set
Consider the following van der Pol–like system presented in (Prajna et al., 2004):
The origin is the unique equilibrium point. The Lyapunov function given by
proves that the origin is globally asymptotically stable, since
Let
Set
since Eq. (6) holds if Eq. (18) holds. Let its boundary corresponds to the “inner” boundary of

For X1(0) = −1.3 and X2(0) = 1.3, the figure shows numerical realizations of Eq. (17). The dynamics of the process are shown in blue. The outer black circle corresponds to
3.1.2. Model-Testing
Consider the van der Pol system:
The origin is the only equilibrium point and globally asymptotically stable. This can be shown by means of the Lyapunov function given by
Assume that Eq. (17) is the system under investigation that we observe. For 20 times 100 realizations of Eq. (17) with X1(0) = 2.8, X2(0) = −2 and stopped if either
and φ = 2.411. By Theorem 1, this implies that Eq. (20) can be invalidated as a model for system Eq. (17). Indeed,
where
3.2. Parametrically different three-dimensional systems
Consider the lorenz-like system
where d is a positive constant. The unique equilibrium point is the origin, whose stability can be shown via
Let
Assume that, for d = 1, Eq. (22) is the system under investigation and the one that we observe. For 20 times 100 realizations of Eq. (22) with d = 1,
and φ = 0.042. We conclude from Theorem 1 that this implies the model with d = 5 represents the data poorly.
3.3. Four-dimensional models for the Hog1 signaling pathway with different noise properties
The MAPK (mitogen activated protein kinase) Hog1 is the most downstream kinase of the signaling cascade, which is activated by osmotic stress (Hohmann et al., 2007). It is also a popular pathway in yeast for benchmark studies. The kinase activity of Hog1 in the cytoplasm leads to the production of glycerol (Westfall et al., 2008), which allows the cells to equilibrate interior and exterior osmotic pressures (Fig. 2). Importantly, this system is adaptive and concentrations of Hog1 quickly return to their preshock levels (Muzzey et al., 2009). This quickly adapting mechanism also allows for “kinetic specificity” such that the slower pheromone pathway is not activated after osmotic shock, albeit sharing some of the pathway components (Behar et al., 2007). The following adaptive model represents the described pathway:
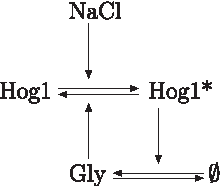
A simple model for Hog1 activation.
Let k1 = 1, k2 = 0.1, k3 = 2, k4 = 3, C1 = 5, C2 = 6, and u = 4. Then, the positive equilibrium point of Eq. (2) is given by
and it is asymptotically stable. Here, x1 denotes concentrations of inactive Hog1, x2 of NaCl, x3 of Glycerol, and C1 the sum of the concentrations of Hog intermediates and NaCl; k1 – k4, u and C2 are positive constants.
The following model assumes noisy total concentrations of Hog1 by setting C1 = X4:
Assume that, for d = 1, Eq. (24) is the system under investigation that we observe. For 20 times 100 simulations of Eq. (24) with d = 1, X1(0) =
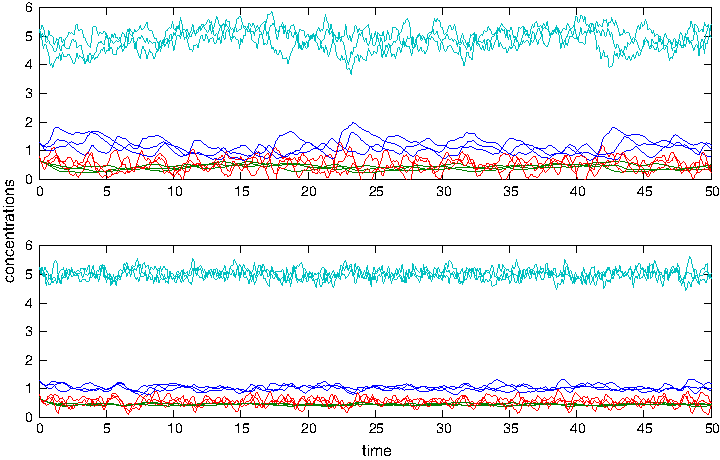
The top panel shows three simulations of Eq. (24) for d = 1 and the lower ones for d = 4, where the dark blue line represents X1 and the others the concentrations of X2, X3, and X4. As expected, noise is much reduced for d = 4.
4. Conclusions
The shortcomings of current experimental design approaches for discriminating between rival mathematical models representing a biological system are that they often use a linearized version of models or cannot deal well with experimental noise and parameter uncertainties. In this article, we presented novel means to computationally test a noisy nonlinear model by comparing it with measurement data of the system. Our approach is based on semidefinite programming techniques, for which efficient solvers exist. Importantly, our method provides analytical results and, thus, avoids extensive simulations. As opposed to analytical methods such as ours, numerical approaches can only approximate the (probabilistic) behavior of the model.
In the Results section, we provided examples of increasing complexity and finally applied our methodology to the Hog1 signaling pathway, a benchmark system, allowing the analysis of noise. Our approach is particularly useful to the field of systems biology, in which research thrives from the interaction between wet-lab work and mathematical modeling, and the models, often derived from data via reverse engineering, try to uncover structure and dynamics of biological systems.
Footnotes
Acknowledgment
This project was financed with a grant from the Swiss SystemsX.ch initiative (grant IPP-2011/115).
Disclosure Statement
No competing financial interests exist.
2
3
However, our approach is not restricted to this type of criteria and allows also the implementation of many other kinds of stopping criteria, which we omit for clarity.