
Abstract
Abstract
Probabilistic approaches for sequence alignment are usually based on pair Hidden Markov Models (HMMs) or Stochastic Context Free Grammars (SCFGs). Recent studies have shown a significant correlation between the content of short indels and their flanking regions, which by definition cannot be modelled by the above two approaches. In this work, we present a context-sensitive indel model based on a pair Tree-Adjoining Grammar (TAG), along with accompanying algorithms for efficient alignment and parameter estimation. The increased precision and statistical power of this model is shown on simulated and real genomic data. As the cost of sequencing plummets, the usefulness of comparative analysis is becoming limited by alignment accuracy rather than data availability. Our results will therefore have an impact on any type of downstream comparative genomics analyses that rely on alignments. Fine-grained studies of small functional regions or disease markers, for example, could be significantly improved by our method. The implementation is available at www.mcb.mcgill.ca/∼blanchem/software.html
1. Introduction
Score-based models of evolution used in sequence alignment are in the process of being replaced by probabilistic models (Lunter et al., 2005; Miklos et al., 2009). Similar transitions have successfully occurred in other areas of bioinformatics such as phylogenetics (Felenstein, 2003), spurred by advantages of using stochastic models that apply equally to sequence alignment. These advantages include more informative parameter estimates, the ability to assess uncertainty in the results, and a neutral model under which tests for selection can be performed. Research on probabilistic indel models and their application to sequence alignment dates back to seminal work by Bishop and Thompson (1986) which was refined by Thorne et al. (1992). Most subsequent work in this field, such as allowing overlapping indels (Miklós et al., 2004), or extensions to multiple alignment (Hein et al., 2003; Holmes and Bruno, 2001), builds on the observation that indel models can be formulated as pair Hidden Markov Models (pair-HMMs) (Durbin et al., 2003). The pair-HMM representation offers the benefit of its accompanying generic algorithms such as Forward, Viterbi, and Baum-Welch, which can be used for alignment and training without the need for model-specific algorithms. It has also led to the use of more general formalisms from linguistics in order to create new, more powerful models. For example, Stochastic Context Free Grammars (SCFGs) have been used to develop a model of evolution where the dependence between nucleotides resulting from pair-bonds in RNA stems could be explicitly modeled (Rivas and Eddy, 2001; Holmes, 2004). Tree-Adjoining Grammars (TAGs) were used to generalize this work to incorporate pseudoknots in the RNA secondary structure (Matsui et al., 2005; Uemura et al., 1999).
One form of inter-site mutational dependence with genome-wide prevalence that is not addressed by any of the current probabilistic models is the relationship between short inserted and deleted sequences and their flanking regions. More specifically, these indels often have a tandem match nearby in the sequence (Fig. 1). Two biological mechanisms, namely replication slippage and unequal crossing over during recombination, are known to cause exactly this phenomenon (Levinson and Gutman, 1987). Recent genome-wide studies have revealed that the vast majority (roughly 90%) of short indels have at least a partial tandem match (Messer and Arndt, 2007; Tanay and Siggia, 2008), making it reasonable to assume that these, or similar, mechanisms are the primary driving force behind the processes of short insertion and deletion and, consequently, evolution in general. Sequence alignment is far from a solved problem (Miklos et al., 2009), so it is logical to seek to use this context signal to improve accuracy through better detection of indels. Some work toward this end has already been done for score-based pairwise alignment, beginning with Benson who extended the Smith-Waterman algorithm to include duplications events which insert an arbitrary number of copies of the repeat sequence (Benson, 1997), increasing the time complexity to O(n4). On sequences with pre-identified tandem repeats, the running time is reduced to O(n2). This problem of aligning sequences of annotated repeats was revisited under a more general model that includes tandem excisions by Bérard and Rivals (2003), who proposed a O(n4) alignment algorithm. Subsequent work allowed for additional events including multiple-excisions, but at the cost of an exponential running time (Sammeth and Stoye, 2006).

Short indels are often flanked by a tandem match. The crossing dependencies shown in the insertion and deletion identified in this sample alignment cannot be modeled by pair-HMMs or SCFGs.
Our aim in this report is to introduce a similar notion of context-sensitive indels into the more modern statistical alignment framework, noting that a similar project was successfully undertaken for context-sensitive substitutions (Siepel and Haussler, 2004). We propose a pair-TAG framework for doing this. In Section 3, we show how certain properties of this model lead to dynamic programming algorithms that are more efficient than previous work using TAGs and even SCFGs. A pairwise aligner based on our model was implemented and tested on a variety of genome sequences, and the increased precision and statistical power of this model are shown on simulated and real genomic data.
2. Pair-TAG Indel Model
We seek to generalize the current HMM-based alignment approach to allow for an indel rate that increases in proportion to the similarity between the inserted or deleted sequence and its flanking sequence as shown in Figure 1. Unfortunately, the “crossing” dependencies between the flanking and indel sites as shown cannot, by definition, be modelled by a HMM or SCFG since languages of the type
A TAG is defined by a quintuple {Σ, NT, Γ, A, S}, where Σ is the set of terminal symbols, NT the non-terminal symbols, and S is the Start symbol. Γ and A are the sets of initial and auxiliary trees respectively. TAG trees are binary, with each node associated with either a terminal or non-terminal symbol from the grammar. All interior nodes of initial trees are labelled with non-terminal nodes, except the root which may be labeled with the Start symbol. All leaf nodes of an auxiliary tree are labelled with terminal nodes except for one designated foot node, which is labeled with the same non-terminal as the tree's root. A TAG combines initial and auxiliary trees to form new trees using two operations: substitution and adjunction. A substitution replaces the leaf of one tree with the root of another while an adjunction operation replaces the internal node of one tree with an auxiliary tree. For examples of these operations, Figure 2. A TAG tree can be mapped to a string by reading the terminal symbols in its leaves in a pre-order traversal. The set of strings generated by a TAG is therefore the set of strings associated with all trees that it can generate.
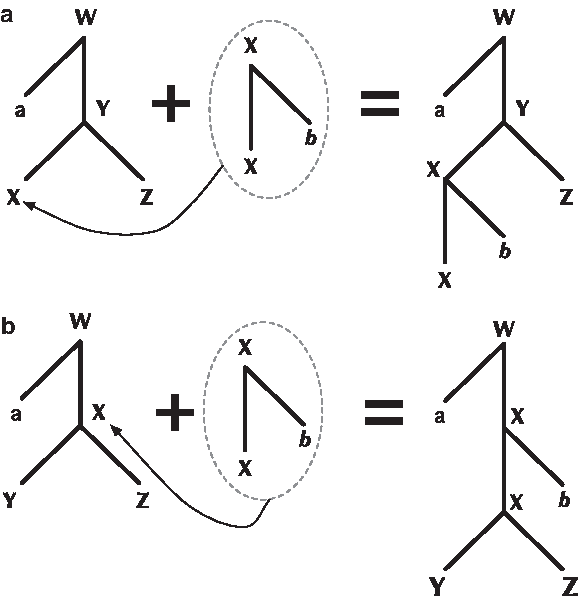
The two TAG operations. (
Substitution, adjunction, and emission probabilities can be added to a TAG in order to generate a stochastic model (Schabes, 1992). Such models have not been widely adopted in practice as TAG parsing algorithms have time complexity O(n6) (Vijay-Shankar and Joshi, 1986), as opposed to the less-prohibitive O(n) and O(n3) of their respective HMM and SCFG counterparts. Still, they have seen some application in bioinformatics. Uemura et al. (1999) developed a model to detect RNA secondary structures including pseudoknots using a custom O(n5) algorithm that was later extended by Matsui et al. (2005) to align a sequence to a known secondary structure. Sequence alignment requires a pair TAG which, analogous to the pair-HMM, would emit a pair of sequences with the corresponding quadratic increase in time complexity (O(n10) for the pseudoknot model (O(n12) in the general case). It is therefore of little surprise that our model, presented below, is, to the best of our knowledge, the first TAG-based sequence aligner. Our approach relies on creating a TAG that is expressive enough to model repeats while still being simple enough to be parsed in an almost-left-to-right manner.
We begin the description of our model by briefly reviewing the pair HMM sequence aligner, which we wish to generalize. A detailed description can be found in Durbin et al. (2003). This model is illustrated in Figure 3, and has a Start state, an End state, and three emitting states: Match, Insert, and Delete. The pair alignment is generated from left to right, with each state emitting a single alignment column. The transition and emission probabilities are derived from reversible indel and substitution models, respectively. The Viterbi algorithm (and traceback) can be used to compute the maximum likelihood (ML) alignment in O(n2). Our air-TAG indel model is illustrated in Figure 4. It is comprised of Start tree, S, and initial trees Γ = {M, I, D, E}, corresponding to Match, Insert, Delete, and End states, respectively. Context sensitive indel operations are supported through the addition of the auxiliary trees,
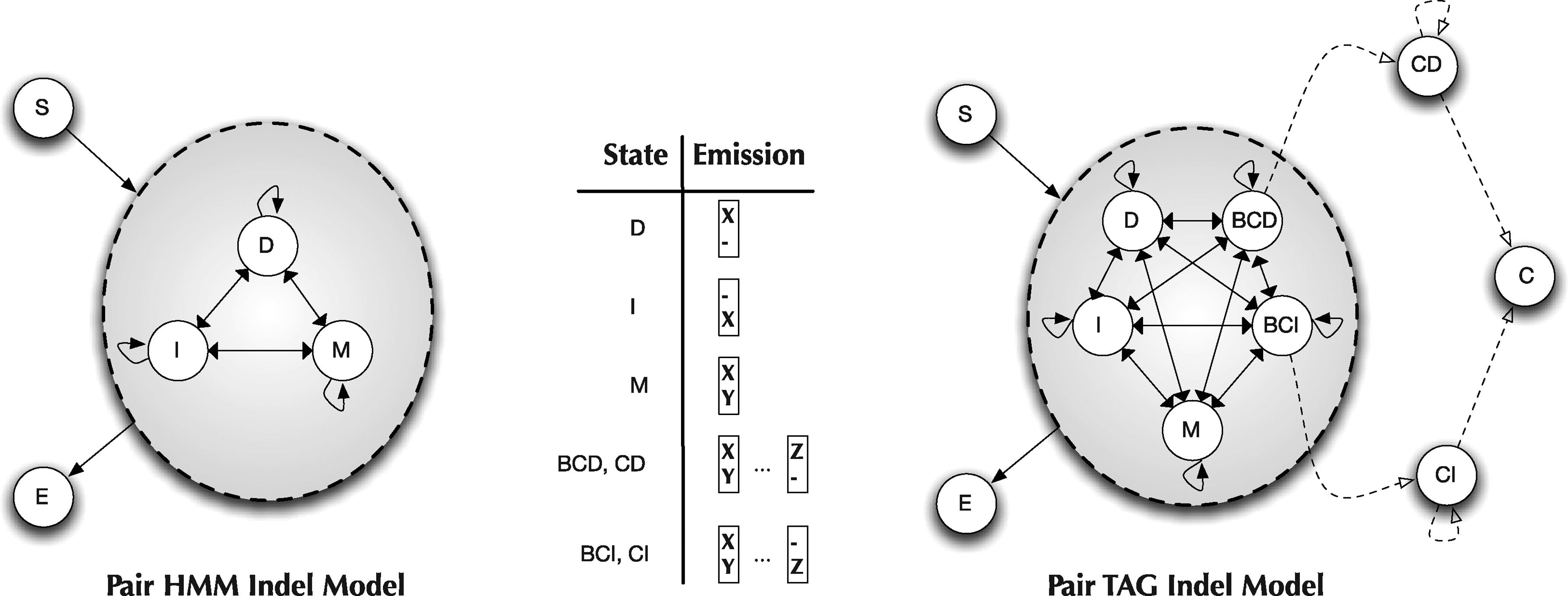
Comparison of the standard pair-HMM model with a HMM-like projection of the TAG model. Substitutions are mapped to solid arrows and adjunctions to dashed arrows. The Emission format for each emitting state is provided, where X, Y, Z are nucleotides.

The pair-TAG indel grammar (
Let sub(X, Y) and adj(X, Y) denote the probability of substituting or adjoining, respectively, tree Y onto tree X. We determine these values using a simple reversible model defined by the following six parameters: Exponential Indel Rate (RI); Geometric Indel Length (PI); Exponential Context Indel Rate (RCI); Geometric Context Indel Length (PCI); Time (t); and Geometric Alignment Length (PA). The substitution probabilities are listed in Table 1, where the function at row i and column j corresponds to sub(i, j). Due to the symmetry of the model, the D and BCD columns need not be shown since, for example sub(I, D) = sub(D, I). All other substitutions, such as sub(I, S), have probability 0. The derivation of sub(M, j), where
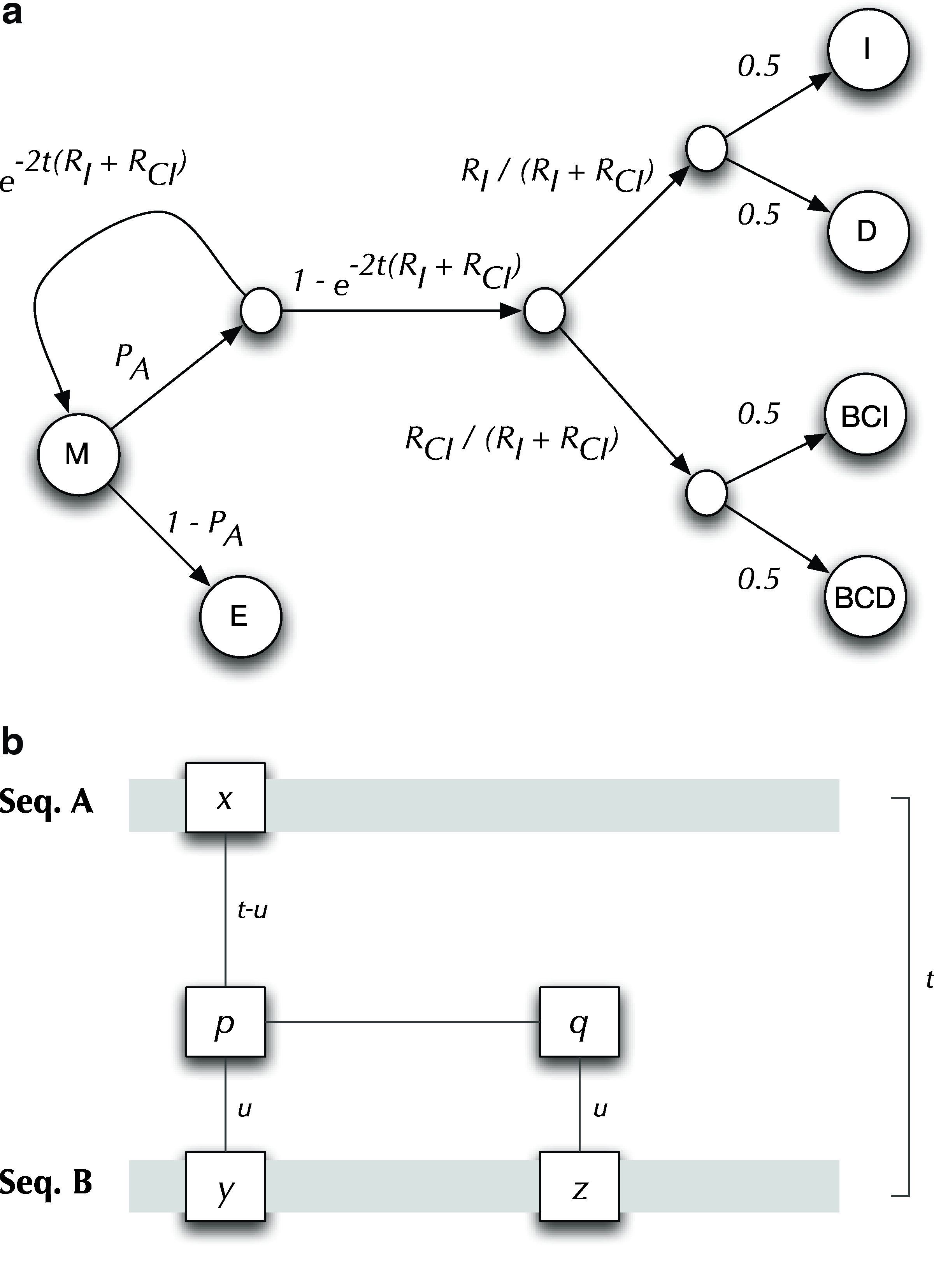
(
Entry at row i and column j corresponds to sub(i, j). In the case where j = E, sub(i, j) = 1 − PA.
We now describe the emission model. Indel (I, D), Match (M) and Context Indel (BCI, BCD, CI, CD) trees emit one, two and three nucleotides, respectively, at their leaves. Emission probabilities are represented using the following notation: emi(x) is the probability an indel emits x; emi(x, y) is the probability a match emits x in sequence 1 and y in sequence 2; and emi(x, y, z) is the probability a context indel emits a match of x, y followed by an indel of z to the right in the alignment. The emission probabilities are derived from a substitution process, sub, and a context dependence dep, both of which can be modeled using standard DNA substitution models. Let Pr sub [x] be the stationary probability of nucelotied x, and Pr sub [y ∣ x, t] the the probability that site with state x mutates into state y after elapsed time t, both according to the substition model sub. It follows that emi(x) = Pr sub [x] and emi(x, y) = Pr sub [x] · Pr sub [y ∣ x, t].
Non-context-dependent emission models would have emi(x, y, z) = emi(x, y) · emi(z). The fundamental contribution of our model is in introducing a dependency between the match and indel sites through the context dependence (dep) process. Let Pr
dep
[y ∣ x] be the probability that a flanking site in a context indel with state x takes on state y in the inserted or deleted string. The substutition and context dependence models can be combined using Equation 1 to compute the distribution of the probability of the indel having occurred via a tandem repeat event. Since the exact time of the indel event is unknown, we must integrate over all time points, and sum over each possible state in the context site (p) and indel site (q) at each time. These variables are illustrated in Figure 5b. The five terms in the equation correspond to branches connecting the five states in the figure.
Equation 1 can be solved analytically using Maple for various types of DNA substitution models such as Jukes-Cantor (Jukes and Cantor, 1969), Kimura (Kimura, 1980), and F84 (Felenstein, 2003) models. The most general combination of models that we could implement and train efficiently in C++ was sub = F84/dep = Kimura. F84 is specified by an arbitrary stationary distribution, and two substitution rates governing transitions and transverstions. Kimura is a restriction of F84 with all stationary probabilities fixed at 0.25. Our context emission model is a function of the following eight (seven free) parameters of the combined F84 substitution and Kimural context dependence models, in addition to time t: Context Transversion Rate (σ); Context Transtition Rate (τ); Substitution Transversion Rate (α); Substitution Transition Rate (β); and Stationary Substitution Distribution (Pr
sub
[x] where
The level of dependence can be tuned using the context parameters σ and τ, from strictly allowing tandem repeats with σ = τ = 0 to full independence as the parameters increase to infinity. We note, finally, that under the F84 model, the transtion rate parameter does not correspond exactly to the transtition rate. It is, rather added to the transversion rate. For example, the rate for A to G transitions is computed as
Figure 6 shows an example of a TAG derivation for a single base insertion followed by a two-base context deletion. An Insert tree is first substituted into the Start tree. A Begin Context Delete tree is then substituted into the resulting tree. The context deletion is continued then closed by adjoining the Context Delete and Close trees, respectively. The process is terminated by substituting the End tree. The resulting alignment is emitted as shown in the figure, and the probability of this alignment is a product of all sub(·), adj(·), and emi(·) terms shown.
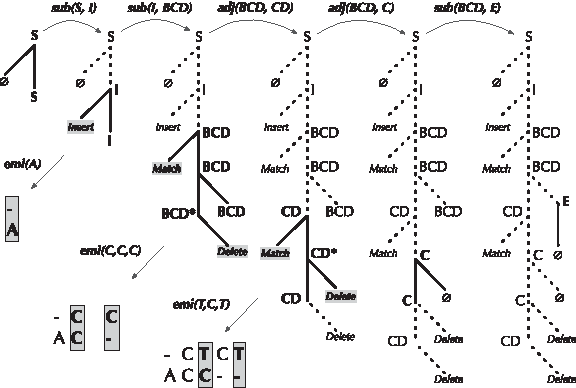
An example of a TAG derivation as it emits an alignment. The derivation tree is illustrated after each of the three substitutions and two adjunctions. The emitted alignment is also displayed in the lower left. The probabiliy of this derivation can be computed as the product of the (eight total) emission, substitution, and adjunciton operations shown.
3. Pairwise Alignment and Parameter Estimation
The most basic question that can be answered using a probabilistic model of evolution is to determine the probability that two sequences are homologous. Algorithmic solutions to this problem can usually be trivially modified to produce ML alignments along with posterior probabilities for alignment columns. Two model properties are often used to speed up these algorithms. The first is reversibility. Felsenstein's pulley principle states that under a reversible model, the probability that sequences A and B are descended from common ancestor C can be computed by assuming one descendant is the ancestor of the other (Felsenstein, 1981). This allows likelihoods to be computed directly, without exploring all possible ancestral states. The second property is Markov dependence, which allows all alignments to be efficiently totaled by the Forward algorithm using dynamic programming (Durbin et al., 2003). An example of a table entry used for this algorithm is illustrated in Figure 7, where F1(i, p) stores the probability that the pair of subsequences formed by the first i characters of A and the first p characters of B are homologous. Markov dependence enables F1(i, p) to be computed from F1(i − 1, p), F1(i, p − 1), and F1(i − 1, p − 1), allowing the table to be quickly filled from left to right. The Markov property applies to pair-HMM models, but is lost when moving to more general formalisms such as pair-SCFGs or pair-TAGs, whose respective parsing algorithms, Inside/CYK (Durbin et al., 2003) and Tag-Parse (Vijay-Shankar and Joshi, 1986) necessitate tables of increasingly higher dimension (Fig. 7). The cost of updating each table entry also increases from O(1) for the Forward algorithm to O(n · m) for the Inside algorithm, and to O(n2 · m2) for TAG-Parse, for a pair of sequences of lengths m and n. Assuming n ≥ m and a constant model size, the space/time complexity of Forward is O(n2)/O(n2), Inside is O(n4)/O(n6), and TAG-Parse is O(n8)/O(n12).
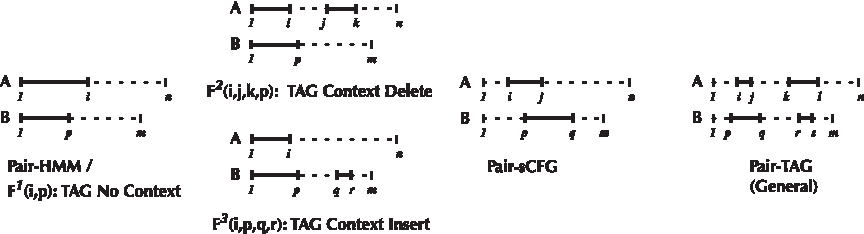
Illustration of the subproblems considered in the dynamic programming algorithms used to parse our TAG Indel model (TAG No Context; TAG Context Delete; TAG Context Insert) along side those required for a pair-HMM, pair-SCFG and general pair-TAG. For each type of model, we show the input strings A and B, the substrings “covered” by each subproblem, and the indices required to denote them.
The TAG Indel model we presented in Figure 4 was designed to mitigate most of the computational overhead, noted above, of moving beyond pair-HMMs. By limiting substitutions and adjunctions to a single point on each tree to the right of previously emitted columns, we ensure a unique “left-to-right” derivation for any alignment. The only exception is columns inserted or deleted by the context operations emitted to the right of adjunction points. These “context windows” are independent since indels do not overlap and can, without any loss of generality, also be derived in a left-to-right fashion. The TAG-Indel derivation can therefore be described as a combination of two different left-to-right processes, context and non-context, linked together in series. These ideas are perhaps best visualized in the HMM-like projection of the TAG-Indel model shown in Figure 3. The black arrows refer to TAG substitutions and the dashed arrows refer to context adjunctions. Alignments are generated as in the pair-HMM by beginning at the S state and then repeatedly transitioning to connected states, emitting columns as specified. When a BCD or BCI state is reached, the model then switches into a context window, transitioning on the dotted connections and simultaneously emitting two columns at a time. The model eventually moves to the C state and reverts back to a non-context mode.
Let s be a TAG Indel tree that is substituted or adjoined in the process of deriving an alignment of sequences A and B. It follows directly from the definition of our model that the range of characters during the derivation up to and including s can be described as one of the three cases illustrated in Figure 7: TAG No Context, TAG Context Insert, or TAG Context Delete. The total probability of all possible alignments of the emitted characters given that s was the last tree used in the derivation is denoted F1(s, i, j), F2(s, i, j, k, p), and F3(s, i, p, q, r) for the three cases respectively. The recursions to compute these values are provided in the Appendix, as is pseudocode for Context-Forward, a O(n4) context-sensitive generalization of the Forward Algorithm. The vast majority of context indels are extremely short, however (Tanay and Siggia, 2008), and by bounding this length with some small constant L, the complexity reduces to O(n2L2). Context-Forward can be modified into Context-Viterbi by changing the summations to max operators, and storing traceback information in addition to likelihoods. The resulting procedure can be used to obtain the ML alignment, or to sample from the ML alignments in a similar manner as is done for HMMs (Durbin et al., 2003). We define the Context-Backward tables, B{1, 2, 3}, as storing the probability that the remaining subsequences of A and B (dashed lines in Fig. 7) are emitted, given that state s was used in the corresponding Forward entry. For example, B2(CD, i, j, k, p) is the probability that the model emits A [i+1, j−1], A[k+1, n] and B[p+1, m] given that it already emitted A[1, i], A[j, k] and B[1, p], and s was the last tree used in the latter derivation. The details of Context-Backward are given in the Appendix. The Context-Forward and Context-Backward tables can be combined to generate ML estimators for the substitution, emission and adjunction probabilities as is done by the Baum-Welch algorithm for HMMs (Durbin et al., 2003). We use these estimators in an Expectation-Maximization (EM) loop to train the model, which is described in full in the Appendix.
4. Results and Discussion
The training and alignment algorithms described above were implemented in C++. The program succeeds at aligning pairs of 100-bp sequences in less than 5 seconds on a typical desktop computer, eventually allowing whole-genome piece-wise realignment on a large cluster in a few hours. We began with a simulation experiment designed to validate the model design and training procedure, as well as the statistical test used later to measure goodness of fit to real data. We simulated a set of 250 pairwise alignments using our model for each possible parameter assignment (144 total) from the following values, limiting context indel lengths to ten to speed up training:
These values were chosen in an attempt to represent those that could be encountered when aligning human to species as close as chimpanzee and distant as mouse. Both the substitution and context models are effectively restricted to Jukes-Cantor models by setting the flat stationary distribution and transition rate parameters to 0. This was done to reduce the number of parameter combinations to a managable number (144). t = 1 was used for all tests, as time cannot be estimated independently from the rates. For each parameter assignment, both the HMM and TAG indel models were trained on the generated data. The estimated parameters were accurate to within a few percent of the true parameters in most cases. There were exceptions in pathological cases, such as {RI = 0.5, RCI = β = τ = 0.01} where the estimate of β was completely wrong (high, in order to compensate for the relatively high indel rates) but these cases are unrealistic, and as liable to fool other models.
To show that estimating these context indel parameters is meaningful, we contrasted the fit of the TAG indel model to that of the pair-HMM using two goodness of fit tests: the Likelihood Ratio Test (LRT) and the Bayesian Information Criterion (BIC), both of which correct for the number of free parameters. After training both models on the data as described above, we computed the total log likelihoods of the Viterbi alignments for each parameter set. The LRT showed that the TAG-Indel model fits the data significantly better than the HMM (p < 10−6, data not shown) whenever the rate of context indel is non-trivial (RCI ≥ 0.01). The BIC is a stricter test that corrects for the data size in addition to assigning an increased penalty for the number free parameters. Under this criterion, the model with the lower BIC value is to be chosen. Again, the TAG Indel model fits the data better under most conditions and as expected, the fit increases in proportion to RCI, RCI/RI, and in inverse proportion to β and τ.
Having validated the training algorithm and goodness of fit test, we proceed to experiments on genomic sequence data. Our model in its current form is not designed to align entire genomes. Rather, we propose it as a method to resolve uncertain gaps within sequences that have at least been roughly aligned. We therefore use short subsequences from BlastZ (Schwartz et al., 2003) pairwise alignments of the human genome to other species as downloaded from the UCSC Genome Browser (Rhead et al., 2009). The subsequences were sampled as follows. The chromosome 22 alignment (.axt file) was divided into blocks of length 60 (including gaps), and 5000 blocks were chosen at random. 1 Implementation improvements such as banding [Hein et al., 2000] and faster numeric estimation procedure could be used to vastly increase the sample lengths in the future. The reference alignments between human (hg19) and chimp (panTro2), gorilla (gorGor1), orangutan (ponAbe2), rhesus macaque (rheMac2), marmoset (calJac2), tarsier (tarSyr1), dog (canFam2), and mouse (mm9), were sampled in this way. Gaps were then removed from each BlastZ alignment block, and the TAG indel model was trained on each sample.
The estimated parameters of the model for the alignments between human and the selected species are given in Table 2. In each case the context indel rate was much higher than the non-context rate, supporting the hypothesis that most short indels are the result of tandem events. The relative rate of context events, RCI/RI, does lower as the species become more diverged and mutations accumulate to prevent the identification of some tandem events. Even though the two-parameter (σ and τ) Kimura model was used for the context dependency, the σ values were near-0 in most cases. This result indicates that, according to our data, transitions and transversions are equally likeliy between indel sites and their corresponding repeats, a hypothesis supported by the goodnes-of-fit tests below. This is in stark contrast to the substitution model (α and β), which showed overall transtion rates
The full emission model (F84/Kimura) was used.
Table 2 showed parameters estimated by training our full model on data sampled from full genome alignments. We refer to this model as TAG-F84/K: TAG indel model with F84 substitutions and Kimura context dependencies. In order to validate the goodness of fit of our model, we retrained all nine combinations of possible TAG and HMM indel models that can be created using F84, Kimura (K), and Jukes Cantor (JC) substition processes on the same data. These range from the most general, TAG-F84/K, to the most specific, HMM-JC (HMM indel model with Jukes-Cantor substitutions). A validation dataset of the same dimensions as the training data was resampled in the same manner, and realligned using each of the nine configurations. A two-way cross validation was then used to test the relative goodness-of-fit of each mode on each species. The results are reported in Figure 8, which displays the difference between the BIC score for each model and that for the best (minimum BIC) model. If one model has a lower bar in the graph than another, it fits the data for that speices significantly better by the BIC test. There is a clear signal across all species, showing that TAG models to fit significantly better than the HMMs. Similarly, this data shows that more general substitution models are to be preferred, however the Jukes-Cantor context model performed better than the Kimura. The latter result is fitting with the negligible σ estimates in Table 2. Some of these trends were less clear for mouse, possibly due to overfitting as a result of insufficient training data size, but the best-fitting model remained TAG-F84/JC for each species.
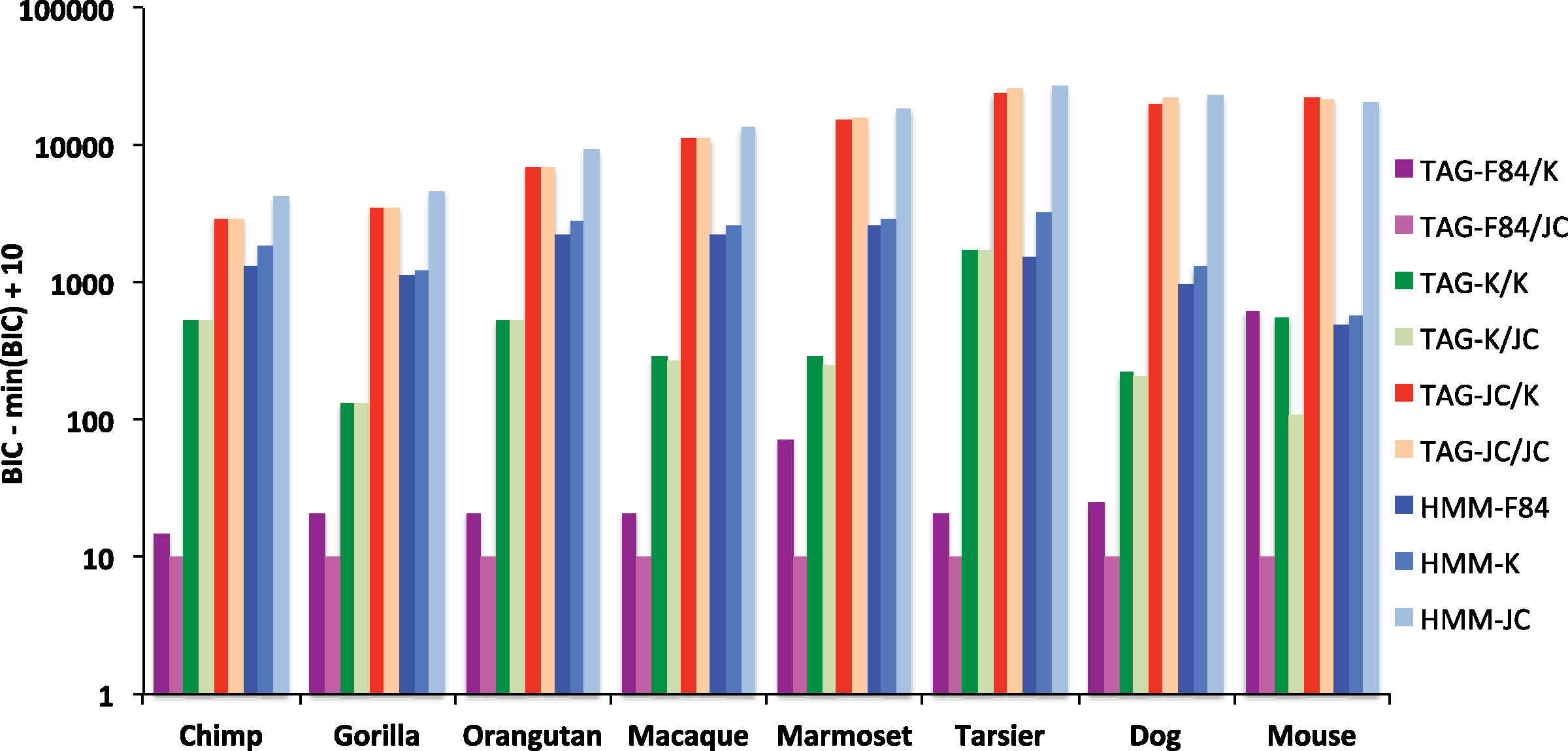
Cross-validation goodness-of-fit results for different parameterizations of the TAG and HMM models. Different combinations of the F84, Kimura (K), and Jukes-Cantor (JC) emission models are presented for the TAG. For example, TAG (F84/K) refers to an emission model comprised of a F84 substitution process and Kimura context dependence. For each speces, the BIC scores for each model are compared to those of the best-fitting model (min(BIC)). If one model has a lower value than another, it is said to fit the data significantly better by the BIC test. The differences are shifted by 10 so that near-zero values are more easily visible.
Finally, we demonstrate how modelling context-sensitive indels improves alignment accuracy. Since the true alignments are not known, we used the parameters estimated from the genomic data to simulate “gold standard” alignments for each organism under the full TAG-F84/K model (Table 2). 1000 pair alignments of length 60 were randomly generated for each species in this manner. A training data set was created by stripping the gaps from these alignments, and all nine model configurations were retrained from scratch on this data. The newly trained models were then used to align the training data, and the results were compared to the gold standard. The TAG and HMM alignment algorithms were designed to always put gaps in the leftmost position possible when breaking ties, so as not to unfairly bias the results (where a systematic error could, for instance, unfairly penalize the HMM for putting indels to the left instead of the right of their tandem matches). The number of alignment errors (pairs of aligned nucleotides present in the gold standard and not in the computed alignment) made made by each method is reported in Figure 9 (TAG-{F84,K,JC}/JC results are omitted as they are almost identical to their respective TAG-{F84,K,JC}/K values). The same trends favoring the more general models in the BIC analysis are apparent in the accuracy results, but the differences between the substitution models are much less significant than between TAG and HMM. Overall, Figure 9 shows that if sequences evolve according to a process with context indel properties (which Fig. 8 supports that it does), then using our pair-TAG aligner over a pair-HMM could result in accuracy improvements, ranging from approximately 5% to 30%. Furthermore, these gains in accuracy are comparable to those obtained when switching to a more general substitution model.
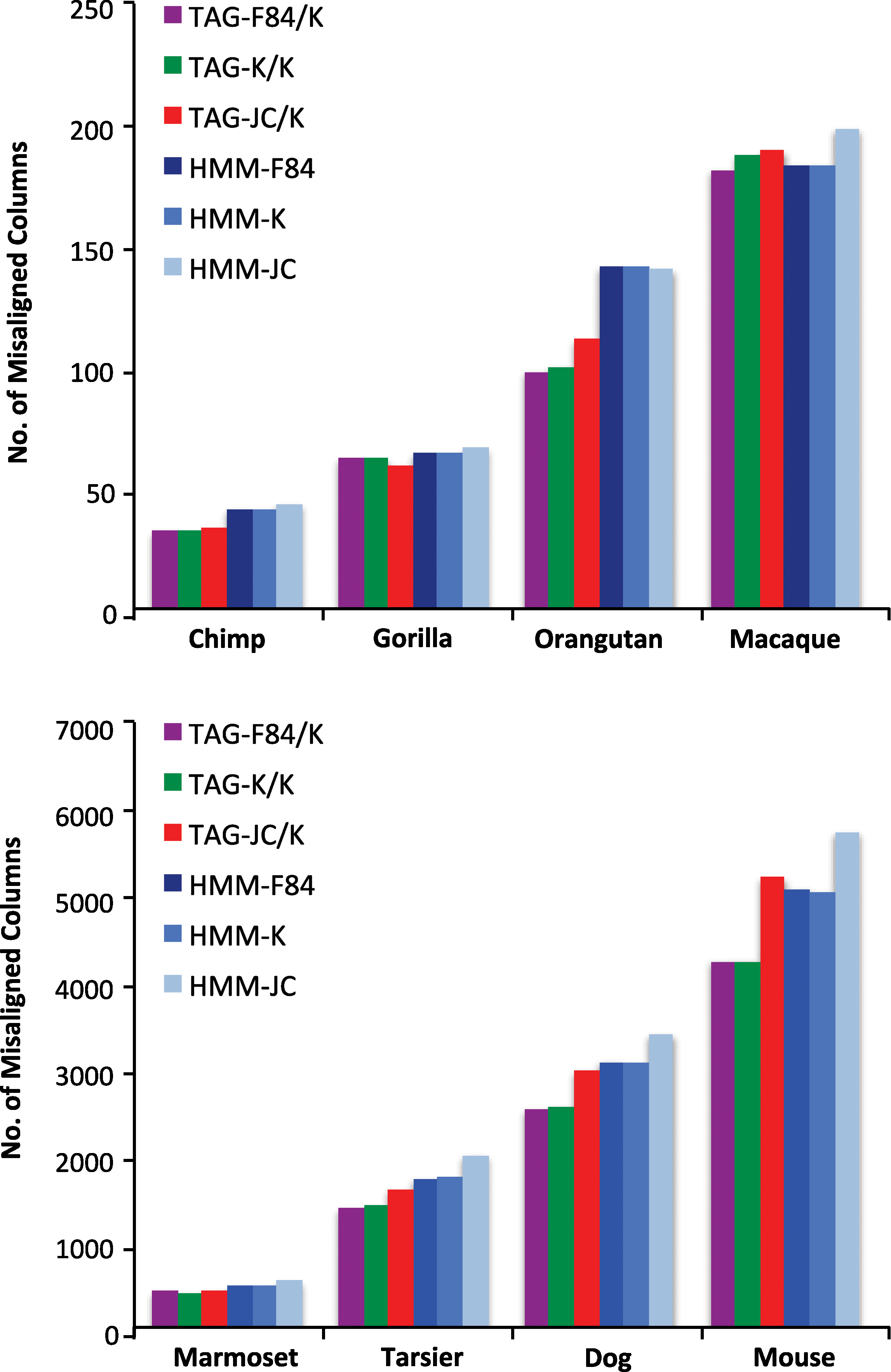
Accuracy results of the different models, based on a comparison with a gold standard simulated under the TAG-F84/K model described in Table 2. The number of misaligned columns is out of roughly 60000.
5. Conclusion
In summary, we developed a probabilistic indel model that generalizes current pair-HMM approaches by adding context-indel events whose probabilities are governed by their similarity to their flanking sequences. This approach was motivated by recent studies showing the prevalence of such events across the human genome, and the fact that many gaps in alignments produced by context-free approaches are still very uncertain, especially in regions exhibiting high divergence. We therefore claim that the accuracy of current methods can be improved by taking into account this context signal. As probabilistic models based on pair-HMMs are being increasingly favored over score-based approaches, we elected to generalize the pair-HMM using a pair-TAG to include context sensitivity. Our simulation experiments demonstrated that our TAG indel model can accurately detect context-sensitive indels and their associated parameters from sequence data. We also showed that it fits pair sequence data from alignments between human and species ranging from chimpanzee to mouse significantly better than a pair-HMM. The estimated rates from these alignments were used to generate “gold-standard” alignments, which the TAG model was able to more accurately recover than the pair-HMM as divergence increased. We therefore conclude that our TAG indel model provides a significant improvement over current methods, and has demonstrated the potential to be used to refine uncertain regions in existing alignments, especially in diverged sequences. These results will have an impact on any analyses that rely on alignments, such as fine-grained studies of small functional regions or disease markers. In the future, we are interested in adding more complex events to our model such as indels within or between the context indel and flanking region, scaling the input data sizes, and applying it to multiple alignment and ancestral sequence reconstruction.
6. Appendix
A.1. Context-forward algorithm
The dynamic programming recursions for Context-Forward are listed below.
The first four equations, which update non context entries of the form F1(s, i, p) are computed exactly as in the Forward algorithm. The context entries are similar except an extra two indices are kept for the context indel, and three characters are emitted instead of two or one. Finally, context indels can be “closed” once the inserted or deleted sequence is immediately flanking the current alignment. Pseudocode for the Context-Forward algorithm is shown below.
A.2. Context-backward algorithm
The recursions for computing B{1,2,3} are presented below. Each entry is associated with a state s and a range (ranges and indices are illustrated in 7), and stores the probability that the model emits the subsequences not in the range, given that the last emission inside the range was s.
The pseudocode for Context-Backward is provided. Its running time is identical to Context-Forward.
A.3. Expectation-maximization loop for estimating parameters
Let Φ = (emi(·),sub(·),adj(·)), and Ψ = (λ, γ, RI, RCI, Pi, PCI, PA). The parameters are learned though iterative re-estimation, beginning with a random seed. First the ML estimates of the substitution, adjunction and emission probabilities,
For
For
Footnotes
Acknowledgments
We thank the anonymous reviewers for their suggestsions. Both authors were funded in part by NSERC.
Disclosure Statement
No competing financial interests exist.
1
Alignment blocks with gaps near the block's boundary (gap of length ≥ k within k sites of the boundary) were excluded, to ensure all gaps' contexts are present. We observe that that this procedure will bias the observed indel length distribution (against longer indels), but note that (1) most indels are short enough to not be significantly affected (Chen et al., 2007; Tanay and Siggia, ![]() ), and (2) we are less interested in the absolute rates detected than in the relative context signal and presenting the potential benefits of a proof-of-concept version of our model.
), and (2) we are less interested in the absolute rates detected than in the relative context signal and presenting the potential benefits of a proof-of-concept version of our model.