
Abstract
Abstract
Molecular dynamics (MD) simulations can now predict ms-timescale folding processes of small proteins; however, this presently requires hundreds of thousands of CPU hours and is primarily applicable to short peptides with few long-range interactions. Larger and slower-folding proteins, such as many with extended β-sheet structure, would require orders of magnitude more time and computing resources. Furthermore, when the objective is to determine only which folding events are necessary and limiting, atomistic detail MD simulations can prove unnecessary. Here, we introduce the program tFolder as an efficient method for modelling the folding process of large β-sheet proteins using sequence data alone. To do so, we extend existing ensemble β-sheet prediction techniques, which permitted only a fixed anti-parallel β-barrel shape, with a method that predicts arbitrary β-strand/β-strand orientations and strand-order permutations. By accounting for all partial and final structural states, we can then model the transition from random coil to native state as a Markov process, using a master equation to simulate population dynamics of folding over time. Thus, all putative folding pathways can be energetically scored, including which transitions present the greatest barriers. Since correct folding pathway prediction is likely determined by the accuracy of contact prediction, we demonstrate the accuracy of tFolder to be comparable with state-of-the-art methods designed specifically for the contact prediction problem alone. We validate our method for dynamics prediction by applying it to the folding pathway of the well-studied Protein G. With relatively very little computation time, tFolder is able to reveal critical features of the folding pathways which were only previously observed through time-consuming MD simulations and experimental studies. Such a result greatly expands the number of proteins whose folding pathways can be studied, while the algorithmic integration of ensemble prediction with Markovian dynamics can be applied to many other problems.
1. Introduction
The development of distributed computing technologies has dramatically extended the range of application of MD techniques. For instance, Pande and co-workers achieved a 1.5 millisecond folding simulation of a 39 residue protein NTL9 (Voelz et al., 2010). In spite of this achievement, this strategy still seems limited to small polypeptides (about 50 residues) and, more importantly, requires several months of parallel computing and typically thousands of GPU's.
In this article, we introduce a complete methodology to address these computational complexity limitations. Our approach aims to complement the range of techniques already offered. Unlike MD simulations that use an all-atom description of structures together with a fine-tuned energy force field, here we use a residue-level representation of the structure with a statistical residue contact potentials. This simplification enables us to sample intermediate structures and build a coarse-grained model of the energy landscape and subsequently simulate folding processes.
Since the seminal work of Levitt and Warshel (1975), it is widely acknowledged that simplified representations of protein structures and motions are required to circumvent computational limitations. A conceptual breakthrough came when Amato and co-workers applied motion planning techniques to the protein folding problem (Tapia et al., 2010; Amato and Song, 2002). The method is much faster than classical MD techniques and enables the study of the folding of large proteins. However, this approach does not predict structures; rather, it requires the three-dimensional (3D) structure of the native state to compute potential intermediate structures and unfolding pathways, on which the folding simulations are performed. It follows that the methodology cannot be applied to proteins with unknown structures and cannot be relied upon to study misfolding processes.
In fact, all the methods previously described face a difficulty common with MD: efficient sampling of the conformational landscape. MD algorithms explore the landscape through force-directed local search and progressive modification of the structure. However, the scalability and numerical efficiency when modeling large molecular structures remains problematic, limiting their application to small molecular systems. On the other hand, motion planning algorithms use a 3D structure of the native fold to predict distant structural intermediates. Accordingly, the accuracy of the method can suffer when intermediates sampled are far away from the native state. Recently, Hosur et al. (2011) have combined efficient motion planning techniques with machine-learning to model proteins as an ensemble, but this approach is effective only in the local neighborhood of the input structure.
Such obstacles have been addressed for RNA molecules by the development of structural ensemble prediction algorithms (McCaskill, 1990; Ding and Lawrence, 1999), and the derivation of a finely-tuned energy model based on experimental data (Turner and Mathews, 2010). Combined together, these techniques enable us to compute the RNA secondary structure energy landscapes and sample structures from sequence information alone. Wolfinger et al. (2004) further demonstrated how an RNA energy landscape can be constructed by connecting these samples together and estimating the transition rates between pairs of interconverting states. The resulting ordinary differential equation (ODE) system can be solved to predict and characterize RNA folding pathways. The method has since been improved to analyze the motion of large RNAs (Tang et al., 2008).
In this article, we propose to expand the methodology developed for RNAs to the more complicated case of proteins. First, we design an algorithm to sample the complete conformational landscape of large protein sequences given sequence data alone. Then, we use this sampling algorithm to build a coarse-grain representation of the energy landscape of a protein, from which we construct an ODE system modeling transition rates between folding intermediates that we solve to simulate protein folding.
We choose to address specifically β-sheet structures. The folding of these structures is particularly difficult to simulate. Indeed, β-sheets are stabilized by inter-strand residue interactions, and thus the folding and assembly of these structures is largely influenced by long-range interactions and global conformational rearrangements. For instance, Voeltz et al. (2010) recently showed that the rate-limiting step in the NTL9 fold was beta-sheet hairpin formation.
Since the original work of Mamitsuka and Abe (1994), several groups have proposed models to predict general β-sheets (Chiang et al., 2006; Kato et al., 2009; Tran et al., 2011). However, none of these methods are capable of computing ensembles of β-sheet structures (i.e., perform an exact enumeration of all β-structures without duplicates) and therefore cannot be used to sample the β-sheet energy landscape.
We recently introduced a structural ensemble predictor for transmembrane β-barrel (TMB) proteins (Waldispühl et al., 2008), continuing earlier work on molecular structure modeling (Waldispühl and Steyaert, 2005; Waldispühl et al., 2006). However, TMBs are a special case of β-sheets where each strand pairs with its two sequence neighbors via an anti-parallel interaction (except the “closing” pair which involves the first and last strands). Here, we expand these techniques to allow any β-strand organization in the β-sheet, with parallel and anti-parallel orientations, and enable the sampling of general β-sheets. This algorithm is implemented in the program tFolder.
We use tFolder to sample the β-sheet conformational landscape and build a coarse-grain model of the energy landscape. More specifically, we cluster protein configurations according to contact distance metrics, and associate each cluster with an intermediate folding state. We use the difference between the ensemble free energies of the clusters to compute the transition rates and build an ODE system that models the energy landscape. Finally, we solve this system to estimate the distribution of conformations over folding time.
This methodology reconciles the MD and motion planning approaches for studying folding pathways. Using tFolder, we are now able to simulate in a couple of minutes on a single desktop the folding of large proteins, and to predict the folding pathways (as well as possible misfolding pathways) of proteins with unknown structures. Thus, we are able to provide a broader range of applications, while offering computational efficiency comparable with motion planning techniques. Although we focus on β-sheet proteins, our method in principle could be extended to describe the folding pathways of a wider class of protein structures.
This article is organized as follows. In Section 2, we describe the tFolder algorithm and explain how we construct the coarse grained energy landscape model. Then, in Section 3, we benchmark our methods. First, we evaluate the accuracy of tFolder for simple inter-strand residue contact prediction and show that it performs comparably with more sophisticated techniques specifically designed for this task. Importantly, our contact predictions are not dependent on the separation between the residue indices, which means an improved “very” long-range contact prediction accuracy. Then, we illustrate the insights provided by our methods by analyzing the energy landscape of the extensively studied Protein G. We show that tFolder predicts the correct folding pathways, and interestingly, our simulation reveals a possible off-pathway structure. All these simulations can be performed on query sequences using our program tFolder, available at http://csb.cs.mcgill.ca/tFolder.
2. Methods
To predict realistic protein folding pathways, we exploit well-established ensemble prediction algorithms (Waldispühl et al., 2008) for their ability to accurately predict the energy scores of millions of feasible structural conformations from sequence alone. Our approach proceeds in two steps: (1) Given an arbitrary peptide sequence, we produce ensemble predictions of the energetic weight for all possible β-sheet structures and sub-structures, utilizing an enhancement to standard ensemble predictors which allows permutation. (2) Using each conformation's energetic score and metrics of conformational similarity, we derive the likelihood of dynamic state-to-state transitions and assemble a set of complete folding paths. In this way, we can identify and rank the most likely pathways from an unfolded conformation to a fully folded conformation based on predicted energy landscapes.
Modeling β-sheet ensembles
We model the set of all possible β-sheet conformations a peptide can attain using a statistical-mechanical framework. Conceptually, each structure is described by the set of residue/residue contacts that form hydrogen bonds between β-strand backbones, and is assigned a Boltzmann-distributed pseudo-energy, determined by the specific residues involved in contacts. To characterize the energetic landscape of this ensemble, a partition function Z can be calculated over all structural states
with energies
Our energy model is based on statistical potentials and follows directly from prior prediction tools that have been shown to be accurate (Waldispühl et al., 2008; Cowen et al., 2001). An energy Ei,j is given to each residue/residue pair within the β-sheet fold following Ei,j = −RT[log(p(i, j)) − Zc], where Zc is a statistical recentering constant and p(i,j) is the probability of these two residues appearing in a β-sheet environment, as observed across all non-sequence-homologous solved structures in the PDB (Waldispühl et al., 2008). Further, we assign separate probabilities based on the hydrophobicity of the environment on either face of a β-sheet.
A naive approach to computing the partition function would thus be to enumerate all possible structures and compute each structure's contribution to the sum individually. However, as was previously shown for the special case of anti-parallel β-strands in transmembrane β-barrel proteins, a much more efficient method exists using dynamic programming (Waldispühl et al., 2008). We have generalized this approach to enable the computation of arbitrary single β-sheet fold topologies.
Permutable β-templates
We introduce the concept of permutable β-templates to enable the calculation of the partition function of a β-sheet with arbitrary β-strand topologies. This extends existing ensemble prediction techniques by allowing any combination of parallel and anti-parallel β-strands to be including within a single β-sheet fold, and by removing any sequence dependency between β-strand /β-strand pairing partners. Prior methods supported only all-anti-parallel β-strands and required β-strand /β-strand interactions to be separated only by coil (and not other strands) (Waldispühl et al., 2008).
To efficiently encode these generic shapes, each strand is labeled

An illustration of how a permutable β-template can be encoded as a signed permutation. The permutation lists the strands in the order that they occur in the sheet, with the sign indicating whether the strand is parallel (+) or anti-parallel (−) to the first strand.
The energy of a structure with n strands, can be recursively defined as E(Sn) = E(Sn−1) + Pairing(sn−1,sn), where E(Sn−1) is the interaction energy between the first n − 1 strands, and Pairing(sn−1,sn) is the energy of the pairing of strand n − 1 with strand n (Fig. 2a). tFolder exploits the shared structure between instances in the ensemble by computing this recursion using a dynamic programming algorithm. The result of each recursive call is stored in a table indexed by the parameters of the call. Subsequent recursive calls made with the same parameters perform a table lookup instead of re-computing the value of the recursion.
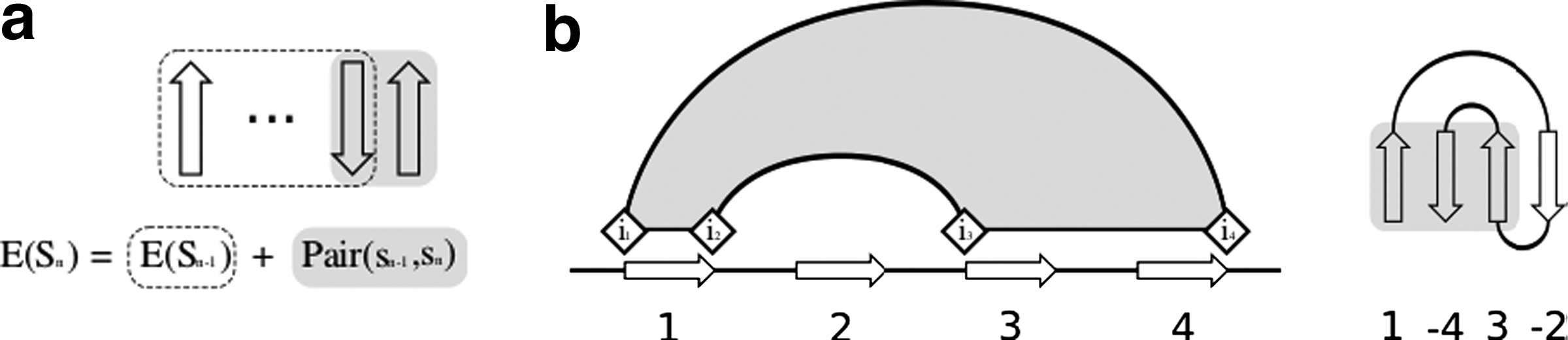
(
For a sheet of n strands, the table has n rows, where the kth row has entries corresponding to valid configurations of the first k strands. For the kth strand, these configurations are partitioned by the location of four indices k1, k2, k3, k4, which denote the boundaries of the region occupied by the k strands (Fig. 2b). To begin, the algorithm enumerates all possible positions of the first two strands, and for each stores the strand pair interaction energy in entry E21222324 of the table. For each subsequent strand k, the value of Ek1k2k3k4 is computed as:
where i1, i2, i3, i4 are enumerated for all valid settings for the boundaries of the preceding strands, given the boundaries of the kth strand. Once the recursion has filled the table, the partition function Z is calculated by summing over all possible settings of n1, n2, n3, n4:
The table constructed to calculate the partition function can be used to sample the distribution of configurations of a given topology, utilizing the approach established by Ding and Lawrence (2003) for RNA secondary structure, and successfully applied previously by Waldispühl et al. (2006) to sample conformations of β-barrel proteins. To do this, we perform a traceback through the table and, at ith step, sample the indices within which the first i strands are contained, according to the Boltzmann representation of these i-stranded structures (Fig. 3).
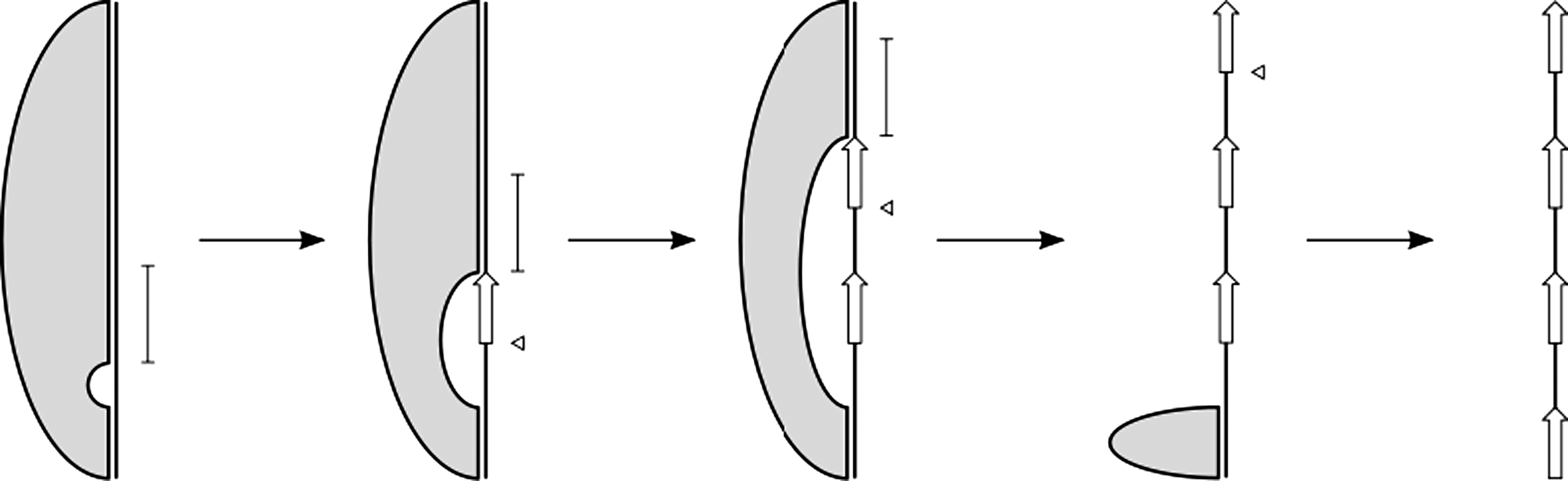
Illustration of of how the sampling procedure performs a traceback through the table, over the indices of intermediate structures. During each step of the sampling procedure, the location of a single strand is sampled from the region indicated by the vertical bars. The triangles denote the location of the strand sampled during the previous step.
2.1. Predicting folding dynamics
Conceptually, we model the folding process as a path through a graph of varyingly folded conformations of a protein. In this graph, different protein conformations are represented as states, and two states that inter-convert in a folding pathway are connected by an edge, analogous to work with RNA described previously (Wolfinger et al., 2004). The tFolder algorithm provides a means to efficiently sample the energetically accessible conformations that make up the states of this graph. We further propose a means to determine the connectivity between states and demonstrate how this can be applied to calculate the dynamics of the folding process.
Since we do not know the final structure, we begin by sampling configurations from all possible permutations of β-sheet topology, as described above. For every pair of states, we add an edge between two states if (1) the states have compatible topologies, and further, (2) the states show structural similarity.
Two templates are compatible if they are identical to each other, modulo the addition or removal of a single strand pairing. This operation can result in the growth of a core structure, or the nucleation of an independent strand pair (Fig. 4). Note that the requirements for satisfying the second criterion of structural similarity depends on the metric used to estimate structural similarity between two conformations. In practice, we use a contact based metric and deem two structures to be structurally similar if the metric is below the transition threshold.
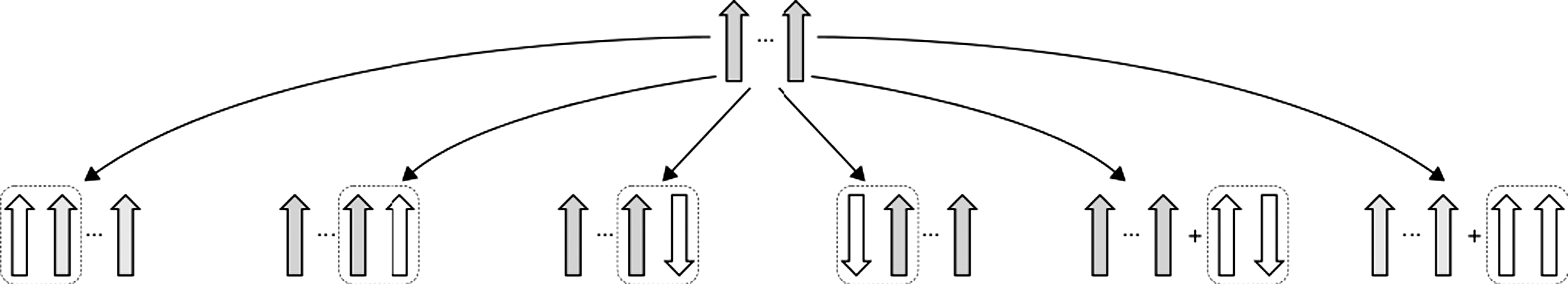
The topologies that are compatible with a given state (shaded gray) result from the addition of a single pairing between strands (dashed box). The “+” indicates that there is no pairing between the gray structure and the white strand pair.
Given the graph constructed according to these two criteria, the change in the probability of the system being in state i at time t is calculated from the total flux into and out of state i,
where pi is the probability of state i, X is the state space, and rij is the rate of transition from state i to state j. Given that two states are connected in the graph, the rate at which two states inter-convert is proportional to the difference between free energies of the states (ΔG); the system tends toward energetically favorable states. We calculate the transition rate rij between states i and j using the Kawasaki rule (with parameter r0 to scale the time dimension):
The dynamics of the system are calculated by treating the folding process as a continuous time discrete state Markov process. Given the matrix of folding rates R, where Rij = rij and initial state density
Since we sample hundreds of states from each β-strand topology, we partition the state space into macro states using clustering, in order to work with a tractably sized system. Under this approximation, we consider two clusters the graph to be connected if the minimum distance between any two states from each cluster is below the transition threshold. We define the ensemble free energy difference ΔGij between two macrostates i and j by summing over the states from which they are composed.
Although this approximation lessens the computational burden, it represents a trade off. The granularity achievable by our simulation is at the level of the macrostates. Note, energy barriers are not explicitly incorporated into the model, since entire β-strands are either added or removed between states without partially-formed intermediates.
3. Results
3.1. Evaluation of contact prediction
To evaluate the contact prediction performance of tFolder, we tested it using a 16-protein benchmark.
1
Proteins were selected from the Protein Data Bank which had 4–6 β-strands with dominantly beta structure, 100 residues or smaller, and sequence identity less than 30%. To ensure a fair comparison with the BETApro and SVMcon, we removed from this set those proteins that were used in the training sets of these methods. From each of these 16 proteins, the β-topology was extracted and used as input for tFolder, along with the amino-acid sequence and fixed strand length of 5–8 residues. Since predicting folding dynamics involves a permutation over all β-topologies, this demonstrates the expected accuracy of each folding state along the pathway. We sample 500 configurations of each protein, and use these ensembles to compute a stochastic contact map and distribution of strand locations (Fig. 5a, and b). The contact map represents the probability of observing a given contact, and predicted contacts are the set of all contacts with probability above a threshold value t. The selection of t influences the measured performance (Fig. 5c), so to objectively set the threshold, we chose a t that maximizes the F-measure. We evaluated the quality of our contact maps based on the Accuracy
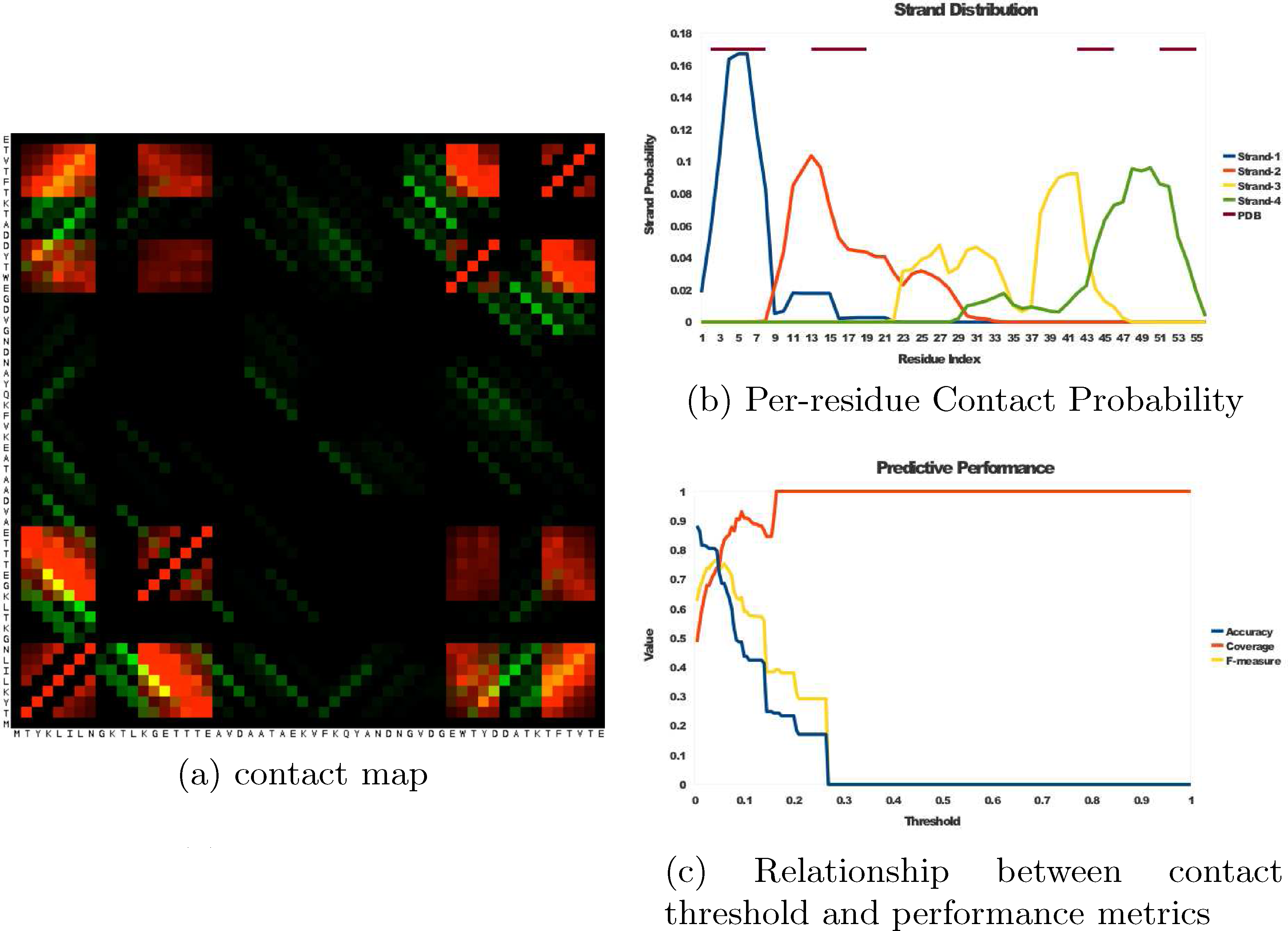
Summary of the distribution of structures predicted by tFolder for Protein G. (
These performance metrics are reported for contacts that are more than 0, 12, and 24 residues apart, showing that tFolder maintains reasonable predictive accuracy even with large contact separations.
In order to evaluate the performance of tFolder with respect to other approaches for contact prediction, we present in Table 2 a comparison with two leading contact prediction algorithms, SVMcon and BETApro. It can be seen that tFolder is able to perform comparably at the task of contact prediction, and performs particularly well for contacts with sequence separation greater than 24 residues. Although these methods sometimes outperform tFolder, it is important to note that the accuracy of tFolder is less sensitive to the distance of contact separation. Since critical protein folding steps can involve both short-range and long-range β-sheet contacts, it is especially important for long-range contacts to be predicted correctly to allow an accurate folding pathways to be reconstructed.
The methods are evaluated based on their ability to perform contact prediction for contacts greater than 12 and 24 residue separation respectively.
3.2. Predicting the folding pathways of the B1 domain of Protein G
To demonstrate the efficacy of our techniques for predicting protein folding pathways, we reconstruct the folding landscape of the B1 domain of Protein G—a well-studied protein for which the pathway has been elucidated through many experimental studies and MD simulations. To do this, all possible permutations of a 4-strand β-sheet topology were sampled and clustered. For each of these sets of structures, the cluster with the highest probability of being observed was selected to be representative of each topology.
The graph of the folding pathway was constructed by considering all pairs of clusters. If the minimum distance beween two clusters was less than a predetermined transition threshold, we considered that there was exchange between the two states. We tried several metrics, including segment overlap, mountain metric, and a contact based metric (Zemla et al., 1999; Moulton et al., 2000), selecting the contact-based metric, because it performed best empirically. The resulting graph of protein conformations is illustrated in Figure 6a. Inspection of this graph, along with the folding dynamics computed from this graph in Figure 6b, reveals folding intermediates consistent with those previously reported by Song et al. (2003). It should also be noted that although we compute other configurations of the sequence that are energetically favorable (faded states in Fig. 6a), they are not predicted to form because they are unreachable from the unfolded state. Interestingly, a four-stranded off-pathway structure is predicted to form, which has not been observed previously. Furthermore, our results agree with the work of Hubner et al., (2004), who show that the anti-parallel beta-hairpin, predicted to form an interaction between residues 39–44 and 50–55, center around known nucleation points W43, Y50, and F54.

(
3.3. Algorithm running time
The computational bottleneck of our approach is the computation of the partition function of a template. The primary factors influencing this calculation are the length of sequence and the number of strands in the β-topology (the depth of the recursion). The partition function for sequences between 40–130 residues and 4–6 strands was calculated using a single 2.66-GHz processor with 512 MB of RAM. The effect of these two parameters on the computation time is depicted in Figure 7 below. Further, computing the parition function across multiple β-templates is trivially parallelizable. The ability to formulate quick, coarse-grained predictions in a matter of minutes, rather than days of atomistic-detail simulation, is a fundamental benefit of our technique.

The time required to compute the partition function increases with increasing size of amino acid sequence, and number of strands. The time was computed by averaging over n = 3 trials, for sequences of 40–130 residues in length, with 4–6 strands.
4. Discussion
We present tFolder, a novel approach for quickly predicting protein folding pathways through the accurate prediction of the conformational landscape of arbitrary β-sheet proteins. What distinguishes tFolder from other computational approaches that attempt to probe protein folding processes is that tFolder does not require vast computational resources; in fact, it can be ran on a single personal computer. To achieve this performance we use a simplified model for protein folding, allowing us to very rapidly compute a coarse-grained picture of the folding of a protein from sequence information alone. This contrasts with methods that attempt to determine folding mechanisms by trying to unfold proteins from their native state. Such methods require the a priori knowledge of the native structure, and as such are not applicable to study protein sequences with unknown structures. When computing protein folding pathways, our method explores all possible β-sheet configurations, and thus does not face this limitation. Interestingly, this independence from known structures could provide insights into off-pathway kinetics, such as the aggregation of proteins into amyloid structures.
Although tFolder only predicts coarse folding pathway transitions in β-sheet proteins, its strength lies in its ability to quickly separate conformational transitions that are critical to folding from those transitions that could simply result from minor structural fluctuations. This complements the use of MD simulations as the MD can be used to explore the nuanced structural interactions that certainly occur near a transition highlighted by tFolder. Further, although we are able to produce good results using a fairly simplistic energy model, a more complicated formulation, such as one including entropic forces, would clearly improve tFolder's analysis. More advanced heuristics also exist (Tang et al., 2008), which more efficiently extract folding pathway information, which could be applied to tFolder.
Understanding the folding dynamics of β-sheet proteins, especially which β-strand contacts drive folding and conformational stability, could help create better models of hierarchical folding, protein aggregation, and evolutionary pressure. Significant overlap likely exists between many proteins' folding pathways to even permit a classification of common transition elements (Fulton et al., 2005); however, creating such a database would only be possible with sufficiently fast and accurate algorithms. tFolder takes a step toward this end by demonstrating techniques for efficiently predicting ensembles of arbitrary β-sheet proteins, and for combining these predictions to construct accurate protien folding transition landscapes.
5. Appendix
Computing the partition function
For a β-sheet with k strands, and residues labeled
Depending on the permutation, the location of ith strand will occur in one of three regions relative to the boundaries of the occupied structure:
1. In the region of 2. In the region of 3. In the region of
Once the energy of the pairing has been computed, the boundaries of the occupied region are updated depending on which region the strand was added to, and whether further strands will be added in the gaps between strands. For example, in the first column, if there remains a strand to be added in the region
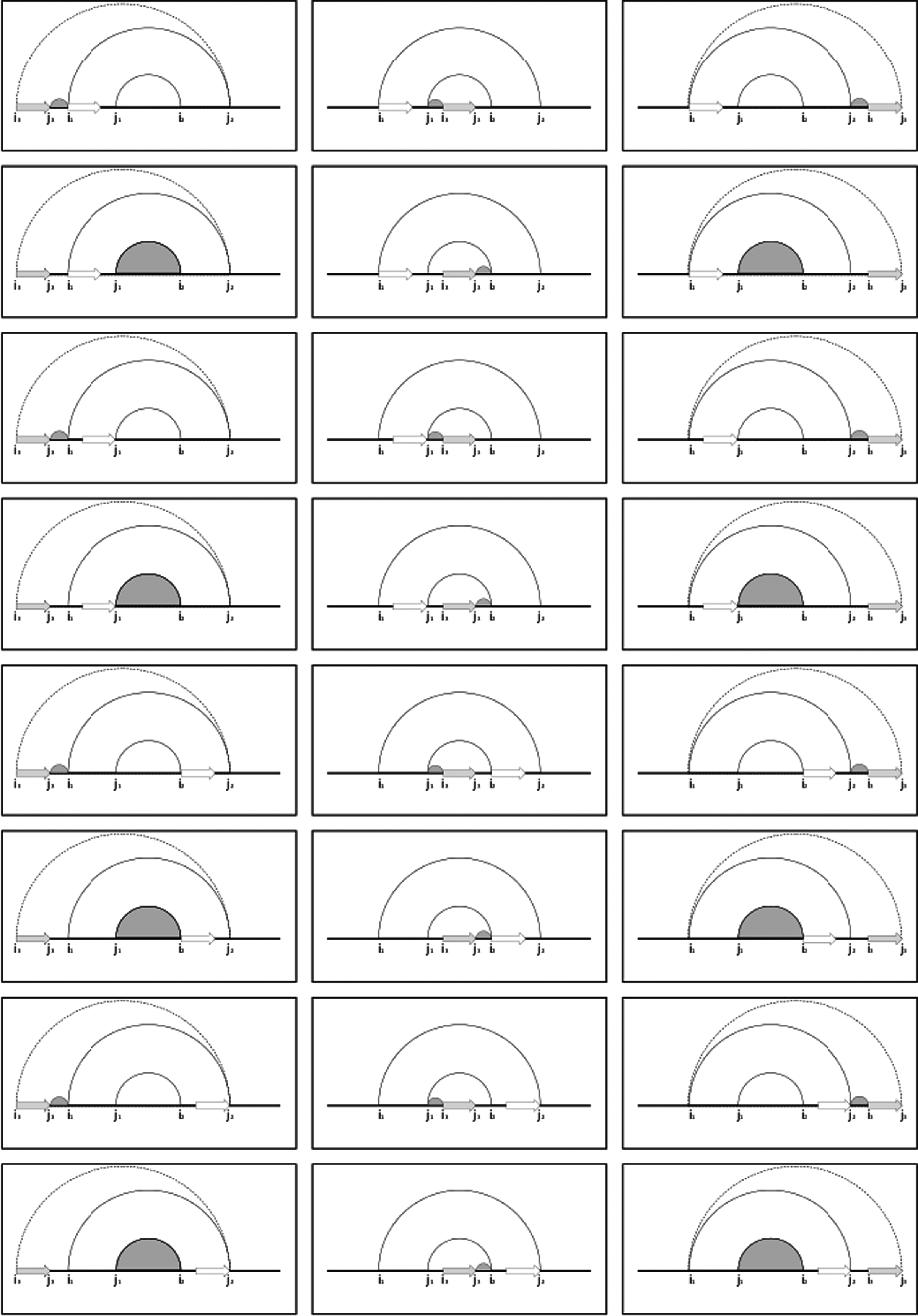
Illustration of the decompositions applied by tFolder used to compute the partition function. The gray arrow represents the addition of the ith strand, and the white arrow is the location of the previous strand, which must be maintained to compute the pairing energy. The solid arcs between (i1, j1) and (i2, j2) represent the boundaries of the previously occupied structure. The dashed arc filled arc represent the updated boundaries after adding the ith strand.

Equations used in the recursive calculation of the folding energy and partition function. The three groups separated by bars represent the three columns in Figure 8.
Footnotes
Disclosure Statement
No competing financial interests exist.
1
Complete benchmark results available at the tFolder website.