
Abstract
Abstract
Current approaches to RNA structure prediction range from physics-based methods, which rely on thousands of experimentally measured thermodynamic parameters, to machine-learning (ML) techniques. While the methods for parameter estimation are successfully shifting toward ML-based approaches, the model parameterizations so far remained fairly constant. We study the potential contribution of increasing the amount of information utilized by RNA folding prediction models to the improvement of their prediction quality. This is achieved by proposing novel models, which refine previous ones by examining more types of structural elements, and larger sequential contexts for these elements. Our proposed fine-grained models are made practical thanks to the availability of large training sets, advances in machine-learning, and recent accelerations to RNA folding algorithms. We show that the application of more detailed models indeed improves prediction quality, while the corresponding running time of the folding algorithm remains fast. An additional important outcome of this experiment is a new RNA folding prediction model (coupled with a freely available implementation), which results in a significantly higher prediction quality than that of previous models. This final model has about 70,000 free parameters, several orders of magnitude more than previous models. Being trained and tested over the same comprehensive data sets, our model achieves a score of 84% according to the F1-measure over correctly-predicted base-pairs (i.e., 16% error rate), compared to the previously best reported score of 70% (i.e., 30% error rate). That is, the new model yields an error reduction of about 50%. Trained models and source code are available at www.cs.bgu.ac.il/∼negevcb/contextfold.
1. Introduction
RNA is typically produced as a single stranded molecule, composed as a sequence of bases of four types, denoted by the letters A, C, G, and U. In addition to the covalent bonds which are formed between adjacent bases in the sequence, every base can form a hydrogen bond with at most one other base, where bases of type C typically pair with bases of type G, A typically pairs with U, and another weaker pairing can occur between G and U. The set of formed base-pairs is called the secondary structure, or the folding of the RNA sequence (Fig. 1), as opposed to the tertiary structure which is the actual three dimensional molecule structure. Paired bases almost always occur in a nested fashion in RNA foldings. A folding that sustains this property is called a pseudoknot-free folding. In the rest of this work, we will consider only pseudoknot-free foldings.
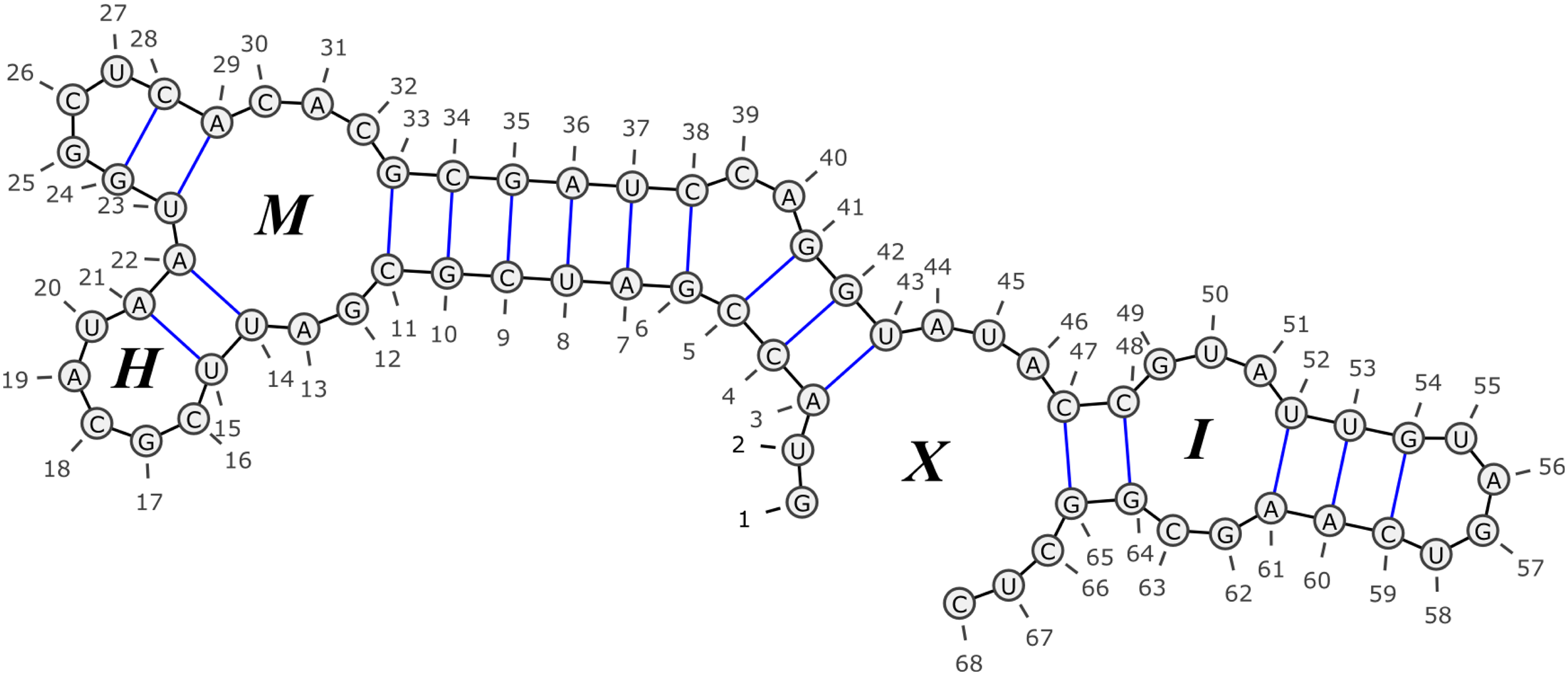
RNA secondary structure. Consecutive bases in the sequence are connected with (short) black edges, where base-pairs appear as blue (longer) edges. The labels within the loops stand for loop types, where H denotes a hairpin, I denotes an internal-loop, M denotes a multi-loop, and X denotes an external-loop. Drawing was made using the VARNA tool (Darty et al. 2009).
RNA structure prediction (henceforth RNA folding) is usually formulated as an optimization problem, where a score is defined for every possible folding of the given RNA sequence, and the predicted folding is one that maximizes this score. While finding a folding that maximizes the score under an arbitrary scoring function is intractable due to the magnitude of the search space, specific classes of scoring functions allow for an efficient solution using dynamic programming (Nussinov and Jacobson, 1980). Thus, in the standard scoring approach, the score assigned to a folding is composed as the sum of scores of local structural elements, where the set of local elements are chosen to allow efficient dynamic programming inference.1
Several scoring models were introduced over the past three decades, where these models mainly differ in the types of structural elements they examine (the feature-set), and the scores they assign to them. A simple example of such a model is the one of Nussinov and Jacobson (1980), which defines a single feature corresponding to a canonical Watson-Crick base-pair in the structure (i.e., base-pairs of the form G-C and A-U, and their respective reversed forms). The score of each occurrence of the feature in the structure is 1, and thus the total score of a folding is simply the number of canonical base-pairs it contains. A more complex model, which is commonly referred to as the Turner99 model, was defined in Mathews et al. (1999b) and is widely used in RNA structure prediction systems (Hofacker et al., 2002; Zuker, 1989, 2003). This model distinguishes between several different types of structural elements corresponding to unpaired bases, base-pairs which participate in different types of loops, loop-length elements, etc. In addition, every structural element can be mapped to one of several features, depending on some sequential context (e.g., the type of nucleotides at base-pair endpoints and within their vicinity), or other values (e.g., the specific loop length, internal-loop asymmetry value).
The parameter values (i.e., scores of each local element) are traditionally obtained from wet-lab experiments (Mathews et al., 1999a), reflecting the thermodynamics free energy theory (Tinoco et al., 1971, 1973). However, the increasing availability of known RNA structures in current RNA databases (Griffiths-Jones et al., 2005) makes it possible to conduct an improved, fine-tuned parameter estimation based on machine-learning (ML) techniques, resulting in higher prediction accuracies. These methods examine large training sets, composed of RNA sequences and their known structures (Sakakibara et al., 1994; Eddy and Durbin, 1994; Dowell and Eddy, 2004; Do et al., 2006, 2007; Andronescu et al., 2007, 2010).
Do et al. (2006) proposed to set the parameters by fitting an SCFG-based conditional log-linear model to maximize the conditional log-likelihood of a set of known structures. The approach was extended in Do et al. (2007) to include automatic tuning of regularization hyperparameters. Andronescu et al. (2007, 2010) used the Turner99 model, and applied Constraint-Generation (CG) and Boltzman-likelihood (BL) methods for the parameters estimation. These methods start with a set of wet-lab parameter values, and refine them using a training set of RNA sequences and their known structures, and an additional data set containing triplets of sequences, structures, and their measured thermodynamic energies. The parameters derived by Andronescu et al. (2010) yield the best published results for RNA folding prediction to date, when tested on a large structural data set.
While the methods for parameter estimation are successfully shifting toward ML-based approaches, the model parameterizations have so far remained fairly constant. Originating from the practice of setting the parameter values using wet-lab measurements, most models to date have relatively few parameters, where each parameter corresponds to the score of one particular local configuration.
We propose a move to much richer parameterizations, which is made possible due to the availability of large training sets (Andronescu et al., 2008) combined with advances in machine-learning (Collins, 2002; Crammer et al., 2006), and supported in practice by recent accelerations to RNA folding algorithms (Wexler et al., 2007; Backofen et al., 2011). The scoring models we apply refine previous models by examining more types of structural elements, and a larger sequential context for these elements. Based on this, similar structural elements could get scored differently in different sequential contexts, and different structural elements may get similar scores in similar sequential contexts.
We base our models on the structural elements defined by the Turner99 model in order to facilitate efficient inference. However, in our models, the score assigned to each structural element is itself composed of the sum of scores of many fine-grained local features that take into account portions of larger structural and sequential context. While previous models usually assign a single score to each element (e.g., the base-pair between positions 5 and 41 in Fig. 1), our models score elements as a sum of scores of various features (e.g., the base-pair (5, 41) has the features of being a right-bulge closing base-pair, participating in a stem, having its left endpoint centering a CCG sequence, starting a CGA sequence, and various other contextual factors).
Our final model has about 70,000 free parameters, several orders of magnitude more than previous models. We show that we are still able to effectively set the parameter values based on several thousands of training examples. Our resulting models yield a significant improvement in the prediction accuracy over the previous best results reported by Andronescu et al. (2010), when trained and tested over the same data sets. Our ContextFold tool, as well as the various trained models, are freely available at www.cs.bgu.ac.il/∼negevcb/contextfold and allow for efficient training and inference. In addition to reproducing the results in this work, it also provides flexible means for further experimenting with different forms of richer parameterizations.
2. Preliminaries and Problem Definition
For an RNA sequence x, denote by Yx the domain of all possible foldings of x. We represent foldings as sets of index-pairs of the form (i, j), i < j, where each pair corresponds to two positions in the sequence such that the bases in these positions are paired. We use the notation (x, y) for a sequence-folding pair, where x is an RNA sequence and y is the folding of x. A scoring model G is a function that assigns real-value scores to sequence-folding pairs (x, y). For a given scoring model G, the RNA folding prediction problem is defined as follows2: given an RNA sequence x, find a folding
Denote by ρ a cost function measuring a distance between two foldings, satisfying ρ(y, y) = 0 for every y and ρ(y, y′) > 0 for every y ≠ y′. This function indicates the cost associated with predicting the structure y′ where the real structure is y. For RNA folding, this cost is usually defined in terms of sensitivity, PPV, and F-measure (see Section 5). Intuitively, a good scoring model G is one such that ρ(y, fG(x)) is small for arbitrary RNA sequences x and their corresponding true foldings y.
In order to allow for efficient computation of fG, the score G(x, y) is usually computed on the basis of various local features of the pair (x, y). These features correspond to some structural elements induced by y, possibly restricted to appear in some specific sequential context in x. An example of such a feature could be the presence of a stem where the first base-pair in the stem is C-G and it is followed by the base-pair A-U. We denote by Φ the set of different features which are considered by the model, where Φ defines a finite number N of such features. The notation Φ(x, y) denotes the feature representation of (x, y), i.e., the collection of occurrences of features from Φ in (x, y). We assume that every occurrence of a feature is assigned a real-value, which reflects the “strength” of the occurrence. For example, we may define a feature corresponding to the interval of unpaired bases within a hairpin, and define that the value of an occurrence of this feature is the log of the interval length. For binary features such as the stem-feature described above, occurrence values are taken to be 1.
In order to score a pair (x, y), we compute scores for feature occurrences in the pair, and sum up these scores. Each feature in Φ is associated with a corresponding score (or a weight), and the score of a specific occurrence of a feature in (x, y) is defined to be the value of the occurrence multiplied by the corresponding feature weight. Φ(x, y) can be represented as a vector of length N, in which the ith entry φ
i
corresponds to the ith feature in Φ. Since the same feature may occur several times in (x, y) (e.g., two occurrences of a stem), the value φ
i
is taken to be the sum of values of the corresponding feature occurrences. Formally, this defines a linear model:
where
The predictive quality of a model of the form GΦ,
3. Feature Representations
We argue that with suitable learning techniques, not being tied to features whose weights could be determined experimentally, and having a large enough set of examples (x, y) such that y is the true folding of x, one could define much richer feature representations than was previously explored, while still allowing efficient inference. These richer representations allow the models to condition on many more fragments of information when scoring the various foldings for a given sequence x, and consequently come up with better predictions. This section describes the types of features incorporated in our models.
All examples in this section refer to the folding depicted in Figure 1, and we assume that the reader is familiar with the standard RNA folding terminology. The considered features broadly resemble those used in the Turner99 model, with some additions and refinements described below, and allow for an efficient Zuker-like dynamic programming folding prediction algorithm (Zuker and Stiegler, 1981). Formal definitions of some of the terms we use, as well as the exact feature representations we apply in the various models, can be found in Section A of the Appendix.
We consider two kinds of features: binary features and real-valued features.
In contrast to most previous models, where each structural element is considered with respect to a single sequential context (and producing exactly one scoring term), in our models the score of a structural element is itself a linear combination of different scores of various (possibly overlapping) pieces of information. For example, a model may contain the features hairpin_base_−1=C_−2=U and hairpin_base_0=G_+1=C_−1=C which will also be generated for the unpaired-base in position 17 (thus differentiating it from the unpaired base at position 25). Note that the appearance of a C-base at relative position −1 appears in both of these features, demonstrating overlapping information regarded by the two features. The decomposition of the sequential context into various overlapping fragments allows us to consider broader and more refined sequential contexts compared to previous models.
The structural information we allow is also more refined than in previous models: we consider properties of the elements, such as loop lengths (e.g., a base-pair which closes a hairpin of length 3 may be scored differently than a base-pair which closes a hairpin of length 4, even if the examined sequential contexts of the two base-pairs are identical), and examine the two orientations of each base-pair (e.g., the base-pair (11, 33) may be considered as a C-G closing base-pair of the multi-loop marked with an M, and it may also be considered as a G-C opening base-pair of the stem that consists of the base-pair (10, 34) in addition to this base-pair). We distinguish between unpaired bases at the “shorter” and “longer” sides of an internal-loop, and distinguish between unpaired bases in external intervals, depending on whether they are at the 5′-end, 3′-end, or neither (i.e., the intervals 1–2, 66–68, and 44–46, respectively). Notably, our refined structural classification allows for the generalization of the concept of “bulges”, where, for example, it is possible to define special internal-loop types such that the left length of the loop is exactly k (up to some predefined maximum value for k), and the right length is at least k, and to assign specific features for unpaired bases and base-pairs which participate in such loops.
Our features provide varied sources of structural and contextual information. We rely on a learning algorithm to come up with suitable weights for all these bits and pieces of information.
4. Learning Algorithm
The learning algorithm we use is inspired by the discriminative structured-prediction learning framework proposed by Collins (2002) for learning in natural language settings, coupled with a passive-aggressive online learning algorithm (Crammer et al., 2006). Algorithms in this class adapt well to large feature sets, do not require the features to be independent, cope well with irrelevant features, and were shown to perform well in numerous natural language settings (Chiang et al., 2009; McDonald et al., 2005; Watanabe et al., 2010). Here, we demonstrate they provide state-of-the-art results also for RNA folding. The learning algorithm is simple to understand and to implement, and has strong formal guarantees. In addition, it considers one training instance (sequence-folding pair) at a time, making it scale linearly in the number of training examples in terms of computation time, and have a memory requirement which depends on the longest training example.
Recall the goal of the learning algorithm: given a feature representation Φ, a folding algorithm
The algorithm works in rounds. Denote by
where:
This is the PA-I update for cost sensitive learning with structured outputs described in Crammer et al. (2006). Loosely, Equation 3 attempts to set
In order to avoid over-fitting of the training data, the final
5. Experiments
In order to test the effect of richer parameterizations on RNA prediction quality, we have conducted five learning experiments with increasingly richer model parameterizations, ranging from 226 active features for the simplest model to about 70,000 features for the most complex one.
5.1. Experiment settings
We then enrich this basic model with varying amounts of structural (St) and contextual (Co) information. St med adds distinction between various kinds of short loops, considers the two orientations of each base-pair, and considers unpaired bases in external intervals, and St high adds further length-based loop type refinements. Similarly, Co med considers also the identities of unpaired bases and the base types of the adjacent pair (i + 1, j − 1) for each base-pair (i, j), while Co high considers also the neighbors of unpaired bases and more configurations of neighbors surrounding a base-pair.
The models St med Co med , St high Co med , St med Co high and St high Co high can potentially induce about 14k, 30k, 86k, and 205k parameters, respectively, but in practice much fewer parameters are assigned non-zero values after training, resulting in effective parameter counts of 4k, 7k, 38k, and 70k. The exact definition of the different structural elements and sequential contexts considered in each model are provided in Section A of the Appendix.
5.2. Results
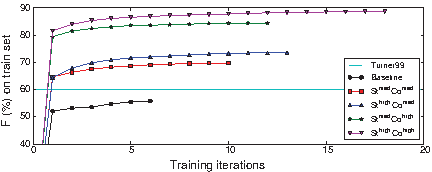
Performance on S-Full-Alg-Train as a function of the number of training iterations.
All models were trained on the training set S-F
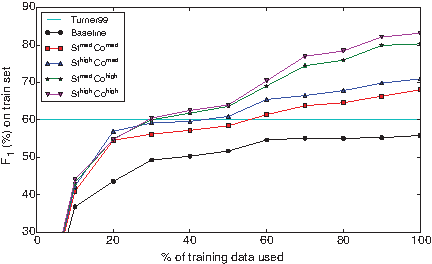
Effect of training set size on validation-set accuracies.
Performance clearly increases as more examples are included in the training. The curve for the Baseline feature-set flattens out at about 60% of the training data, but the curves of the feature-rich models indicate that further improvement is possible with more training data. Thirty percent of the training data is sufficient for St med Co high and St high Co high to achieve the performance of the Turner99 model, and all but the Baseline feature set surpass the Turner99 performance with 60% of the training data.
Table 2 shows the accuracies of the models on the different RNA families appearing in the development set. Interestingly, while the richest St high Co high model achieves the highest scores when averaged over the entire dev set, some families (mostly those of shorter RNA sequences) are better predicted by the simpler St high Co med and St med Co high models. Our machine-learned models significantly outperform the energy-based Turner99 model on all RNA families, where the effect is especially pronounced on the 5S Ribosomal RNA, Transfer RNA and Transfer Messenger RNA families, for which even the relatively simple St med Co med model already outperform the energy-based model by a very wide margin.
Only families with more than 10 examples in the development test set are included. The highest score for each family appears in bold. The CG* and BL-FR* columns were obtained by running the software of Andronescu et al. (2010) with the corresponding models (note that the parameters for these models were obtained by training on the entire S-F
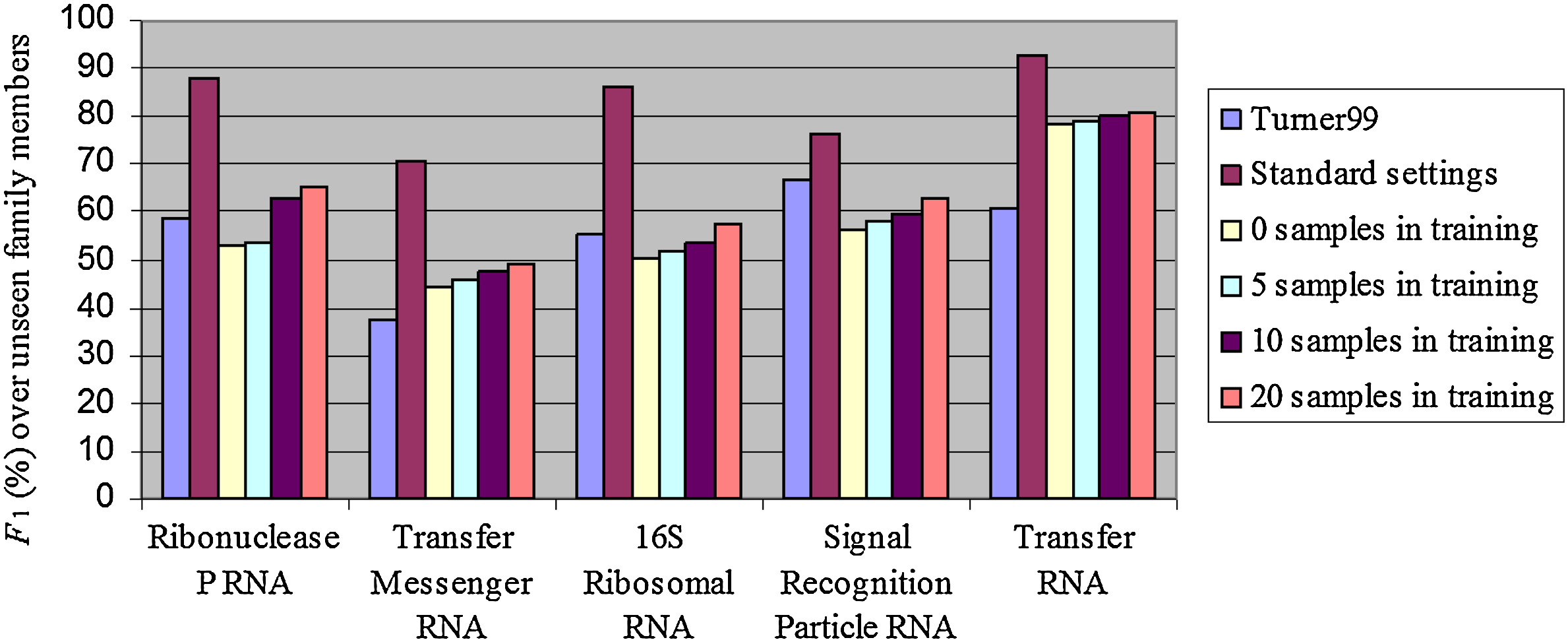
Performance over RNA families which were excluded from the training data. The Turner99 and Standard settings bars give the corresponding family performances in Table 2 of the Turner99 and the StHighCoHigh (trained over S-Full-Alg-Train) models, measured over family members in S-Full-Alg-Val, and are added for reference.
The prediction accuracies over completely unseen RNA types are comparable with those of the Turner99 model, indicating that our algorithm indeed learned general properties of RNA secondary structure. However, results are still considerably lower than those where there is a sufficient coverage of the family in the training set. This may indicate that different RNA families exhibit different features or are folded according to different rules, and that this kind of information may not be transferable across RNA families, at least not when considering the surface sequence alone. However, once the algorithm has seen enough examples of a family in order to effectively learn the parameters governing its folding process, it does a good job at identifying the correct family and folding it accordingly.
Results of previous models are as reported in Andronescu et al. (2010). Models marked with ‡ are trained on S-F
The Baseline model with only 226 parameters achieves scores comparable to those of the Turner-99 model, despite being very simple, learned completely from data and not containing any physics-based measurements. Our simplest feature-rich model, St med Co med , having 4,040 parameters, is already slightly better than all but one of the previously best reported results, where the only better model (BL-FR) being itself a feature-rich model obtained by considering many feature-interactions between the various Truner99 parameters. Adding more features further improves the results, and our richest model, St high Co high , achieves a score of 84.1 F1 on the test set – an absolute improvement of 14.4 F1-points over the previously published best results for this data, amounting to an error reduction of about 50%. Note that the presented numbers reflect the prediction accurcy of the algorithms with respect to a specific dataset. While it is likely that similar accurcy levels would be obtained for new RNAs that belong to RNA families which are covered by the testing data, little can be said about accurcy levels for other RNA families.
6. Discussion
We showed that a move towards richer parameterizations is beneficial to ML-based RNA structure prediction. Indeed, our best model yields an error reduction of 50% from the previously best published results, under the same experimental conditions. Our learning curves relative to the amount of training data indicate that adding more data is likely to increase these already good results. While prediction quality is evidentially higher when predicting foldings of RNA sequences from families that were accessible to the training procedure, it is not inferior with respect to the prediction quality of energy-based models over sequences from unseen families. It is likely that better parameterizations are possible, and the search for a better richly-parameterized model is a fertile ground for exploration.
Our method has some limitations with respect to the physics-based models. In particular, our models scores do not represent free energies. However, there is no reason to use the same model for both structure prediction and free-energy estimation, which can be considered as two independent tasks. Instead, one model can be used for structure-prediction, and the free-energy of the predicted structure can be estimated using a different model which is tailored specifically to the energy-prediction task. Decoupling of structure-prediction and energy estimation is potentially beneficial for both tasks, as there is evidence that the final RNA structures do not neccessarily have the lowest energy among all the possible structures.
Another shortcoming of our current approach is that while it considers a single, margin-based, error-driven parameter estimation method and is optimized to predict one single best structure, it cannot compute the partition function, base-pair binding probabilities and centroid structures derived from them. Probabilistic, marginals-based (i.e., partition function based) training and decoding is an appealing alternative.
Finally, as the learned parameter weights are currently not interpretable, we would like to explore methods of analyzing and trying to make biological sense of them.
7. Appendix
A. Model descriptions
We specify here the features that are used by our models. Each feature description is composed of two parts: a description of a structural element, and a (possibly empty) description of a sequential context. All models discussed in the article are obtained by combining a set of structural elements St with a set of sequential contexts Co, and producing all corresponding features (i.e. producing a feature for each structural element in St and a corresponding sequential context from Co). In Section A.1, we define the different loop types, which are used for categorizing structural elements. In Section A.2, we give the three sets of structural elements St base , St med , and St high , and in Section A.3, we give the three sets of sequential contexts Co base , Co med , and Co high , which are used in our models.
The experiments in the article were conducted with respect to five scoring models: Baseline = St base Co base , St med Co med , St high Co med , St med Co high , and St high Co high . Unpaired base features were obtained by taking each unpaired base structural element in the St set, and combining it with each single position sequential context defined in the Co set. Base-pair features were obtained by taking each base-pair structural element in the St set, and combining it with each double position sequential context defined in the Co set. Length features were obtained only from the St set, as described in Section A.2.
All examples in the text refer to the sequence-folding (x, y) depicted in Figure 1.
A.1 Loop definitions
Let y be a folding (of some RNA sequence x), and let (i, j) be a base-pair in y. Say that a base-pair
A loop is an entity which is composed from the following structural elements:
• Exactly one closing base-pair (i, j). • A set of zero or more base-pairs which are covered directly by (i, j). Such base-pairs are called opening base-pairs of the loop. • A set of zero or more unpaired bases which are covered directly by (i, j).
Loops are traditionally classified into three groups: hairpins (HP) are loops which do not have any opening base-pairs (as the loop closed by (15, 21)), internal-loops (IL) are loops which have a single opening base-pair (as the loop closed by (48, 64) and opened by (52, 61)), and multi-loops (ML) are loops which have more than one opening base-pair (as the loop closed by (11, 33), and opened by (14, 22) and by (23, 29)). Another group of “artificial” loops, which are called external-loops (XL), correspond to sets of elements which are not covered by any base-pair. External-loops may be thought of as loops which are closed by an artificial base-pair between positions 0 and |x| + 1 in the sequence, and may include zero or more opening base-pairs and zero or more unpaired bases (note that each folding of x defines exactly one external-loop, as the one marked with an X in the example; The latter external loop contains the opening base-pairs (3, 43) and (47, 65), and the unpaired base intervals 1–2, 44–46, 66–68).
In order to increase structural element granularity, we refine the hairpin and internal-loop categorization according to their lengths. The length of a hairpin closed by the base-pair (i, j) is defined to be j − i − 1 (i.e., the number of unpaired bases within the loop). An l-HP is the refined classification of all hairpins whose length is l. For example, the hairpin which is closed by the base-pair (15, 21) is a 5-HP. For each set of structural elements St which is defined in the next section, a parameter K was fixed, such that hairpins shorter than K were categorized according to their specific lengths, and hairpins of lengths K or above where categorized as “LONG-HP.”
For an internal loop which is closed by the base-pair (i, j) and is opened by the base-pair (p, q), the left and right lengths of the loop are the values p − i − 1 and j − q − 1, respectively. That is, the left length is the number of unpaired bases between i and p in the loop, and the right length is the number of unpaired bases between q and j. As for hairpins, internal loops are classified according to these lengths, where an “l-r-IL” is a classification of all internal-loops for which the left length is l and the right length is r. For example, the internal-loop closed by (48, 64) in the example (and opened by (52, 61)) is a 3-2-IL. Other traditional loop types such as stems (or stackings) and bulges can be viewed as special cases of internal-loops: a left bulge is an l-0-IL for some l > 0, a right bulge is a 0-r-IL for some r > 0, and a stem is a 0-0-IL. For each set of structural elements St, a parameter I and a series
A.2. Structural elements
Structural elements of a sequence-folding pair (x, y) are inferred solely from the folding y (and the length of x), and do not consider the specific nucleotide composition of x. Each such element which is considered by our models either corresponds to a single base-pair, a single unpaired base, or an interval of unpaired bases. The elements are further categorized according to the loop in which they participate. Recall that y is represented as a set of index-pairs of the form (i, j), 1 ≤ i < j ≤ |x|, where a pair (i, j) in y indicates that the bases at positions i and j of the sequence x are paired in the folding. We say that a base r in x is unpaired in y, to indicate that there is no base-pair
Base-pair elements are classified according to the loop type in which they participate, and whether they open or close this loop. The list of base-pair structural elements is summarized in Table 4.
Unpaired bases are distinguished according to the type of interval of unpaired bases in which they participate. These intervals my be enclosed by a hairpin, an internal-loop, a multi-loop, or an external loop. Internal-loop intervals are further distinguished according to the length of the shorter side of the loop (where there is a unified context for the case when the length of the shorter side is I or longer), and whether they compose the shorter or longer side of the loop. External-loop enclosed intervals are distinguished according to their location in the sequence, which may either be the 5′-end of the sequence, the 3′-end, or neither. Table 5 summarizes the unpaired base types.
The last type of structural elements are unpaired-interval length elements. These elements correspond to intervals of unpaired bases enclosed within hairpins, intervals of unpaired bases in the longer sides of internal loops, and intervals of unpaired bases in external-loops, at their the 5’-end, 3’-end, or neither. For internal-loops, unpaired interval elements are generated only up to some length bound. Each structural element set St defines, in addition to I and the series
Length elements may produce either binary features or real valued features, where no sequential context is examined with respect to such features. Binary features are generated when the length belongs to some length-range related to the feature (e.g., it is possible to generate a binary feature for all hairpin length intervals, whose lengths are 16–20 bases). Real-valued features obtain their value by computing some function of the length. In this work, we use the log function in order to compute occurrence values. For example, it is possible to define a real-valued feature for hairpin length elements, where the values of occurrences of this feature are the log of the corresponding interval lengths. Table 6 summarizes the length structural element types, and their inferred real-valued and binary features. These features were generated for all scoring models we apply (where the set of length elements may differ between the models). A binary feature of the form TYPE-min-max occurs whenever a length interval of type TYPE appears in the structure, and the length of this interval is between min and max (both inclusive). The range sizes used for binary features are 2, 4, 8, and 16, where these ranges start at different offsets. For each range size r, we have produced 100 ranges starting at
In order to explore different resolutions of parameterization, we have defined three sets of structural elements: St
base
, St
med
, and St
high
. These sets differ in the values of the corresponding parameters
A.3 Sequential contexts
Sequential contexts are defined by the notion of context templates. A single position context template T is implemented as a vector of offsets with respect to a given position i in the sequence, e.g., T = [0, −1, 2]. A single position context template generates all possible sequential contexts which may be observed when examining the type of bases at indices which correspond to the specified offsets, with respect to the given position. For example, the context template above will generate the contexts 0=A_−1=A_2=A, 0=A_−1=A_2=C,…,0=U_−1=U_2=U. In order to deal with positions which are at the proximity of the sequence endpoints, we add to the RNA alphabet an additional “out-of-range” character, which is assumed to be observed whenever a context offset points to some index which is smaller than 1 or grater than |x|. Thus, the number of different possible contexts defined by a context template with k offsets is bounded by 5 k (note that not all possible contexts are valid, e.g. the context −1=G_0=out-of-range_1=C).
A double position context template T is defined as a pair of single position context templates T = (T1, T2), and is used to generate sequential contexts of base-pair elements (where T1 defines the sequential contexts of the first base in the pair, and T2 defines those of the second one).
Each one of the context sets Co base , Co med , and Co high corresponds to a set of single position context templates, which generate sequential contexts that are later combined with all unpaired base elements to generate specific model features, and a set of double position context templates, which is used for generating base-pair features. These sets are specified in Table 8. Note that the empty context template [] implies that no sequential context is examined, and therefore when combined with an unpaired base element (such as ML-BASE) yields a feature that only represents the appearance of this element, regardless of any sequential information.
B. Sparsification
The standard approach for implementing folding prediction algorithms is by the application of dynamic programming (Nussinov and Jacobson, 1980; Zuker and Stiegler, 1981). The reader is referred to Durbin et al. (1998) for a simple description of the basic algorithm. In general, such algorithms compute the folding scores of each one of the O(n2) substrings of an input RNA string of length n. For each substring, the algorithm examines certain solutions for smaller substrings in order to compute its score. In terms of theoretical time complexity analysis, the bottleneck part of this computation is induced by the need to examine all possible splits of each substring in some position between its two endpoints (in order to compute multi-loop scores). Since this examination implies that O(n) operations are conducted for each substring, the overall number of operations along the run is O(n3) (except for the split-examination part, there is a constant amount of operations in the score computation of each substring).
The number of examined split points can be reduced by using sparsification techniques (Wexler et al., 2007; Backofen et al., 2011). The underlying observation which is applied in such approaches is that some split points are dominated by others, and their specific examination would not contribute to the calculation. This allows to avoid the examination of a significant portion of split points along the run of the algorithm, while still computing the exact same scores.
Our implementation integrates the sparsification techniques presented in Backofen et al. (2011). In this approach, split points need to be examined only if the prefix of the string which is induced by the split satisfies the so called STEP criteria, and the suffix satisfies the so called OCT criteria (for the formal definition of these terms, see Backofen et al. [2011]). In order to evaluate its effectiveness, we have also run the algorithm under three degenerated sparsification forms: examining split points such that the corresponding suffix satisfies the OCT criteria (without a restriction on the prefix; this is equivalent to the sparsification of Wexler et al. [2007]), examining split points such that the corresponding prefix satisfies the STEP criteria (without a restriction on the suffix), and examining all split points. For each one of the lengths
Figure 5 presents the average amounts of split points per sequence length and sparsification level (denoted by STEP-OCT, OCT, STEP, and NONE, respectively). It can be seen that the number of examined split points when using the OCT and the STEP-OCT sparsification techniques is dramatically lower than in the case where no sparsification is applied. Figure 6 zooms only on the results for the OCT and the STEP-OCT curves, and Figure 7 presents the results in a log-log scale.
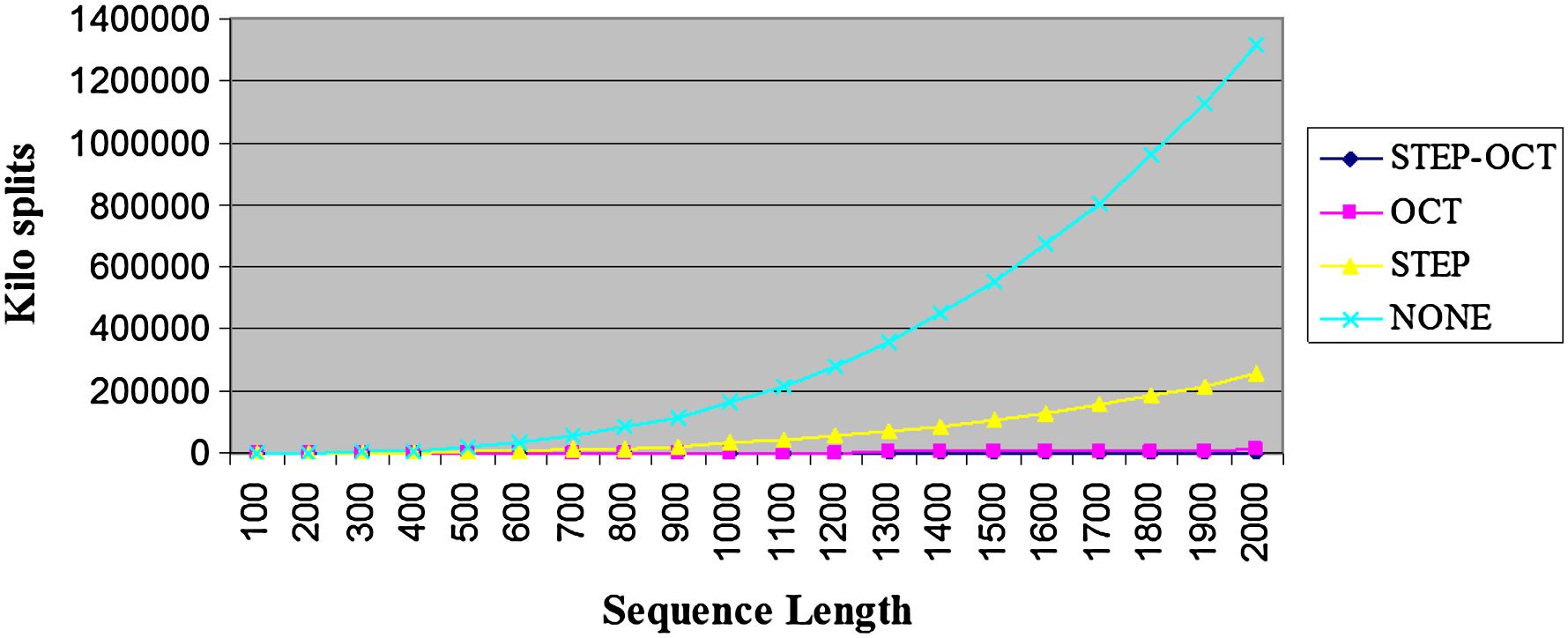
Average examined split points per sequence length. The NONE curve corresponds to the case where no sparsification was applies. The STEP curve corresponds to the case where split points were examined only if the induced prefix satisfies the STEP criteria. The OCT curve corresponds to the case where split points were examined only if the induced suffix satisfies the OCT criteria. The STEP-OCT curve corresponds to the case where split points were examined only if the induced prefix satisfies the STEP criteria, and the induced suffix satisfies the OCT criteria. Since the STEP-OCT curve is of a significantly lower magnitude with respect to the NONE curve, we refer the reader to Figure 6 in which only the STEP-OCT and OCT curves are shown.
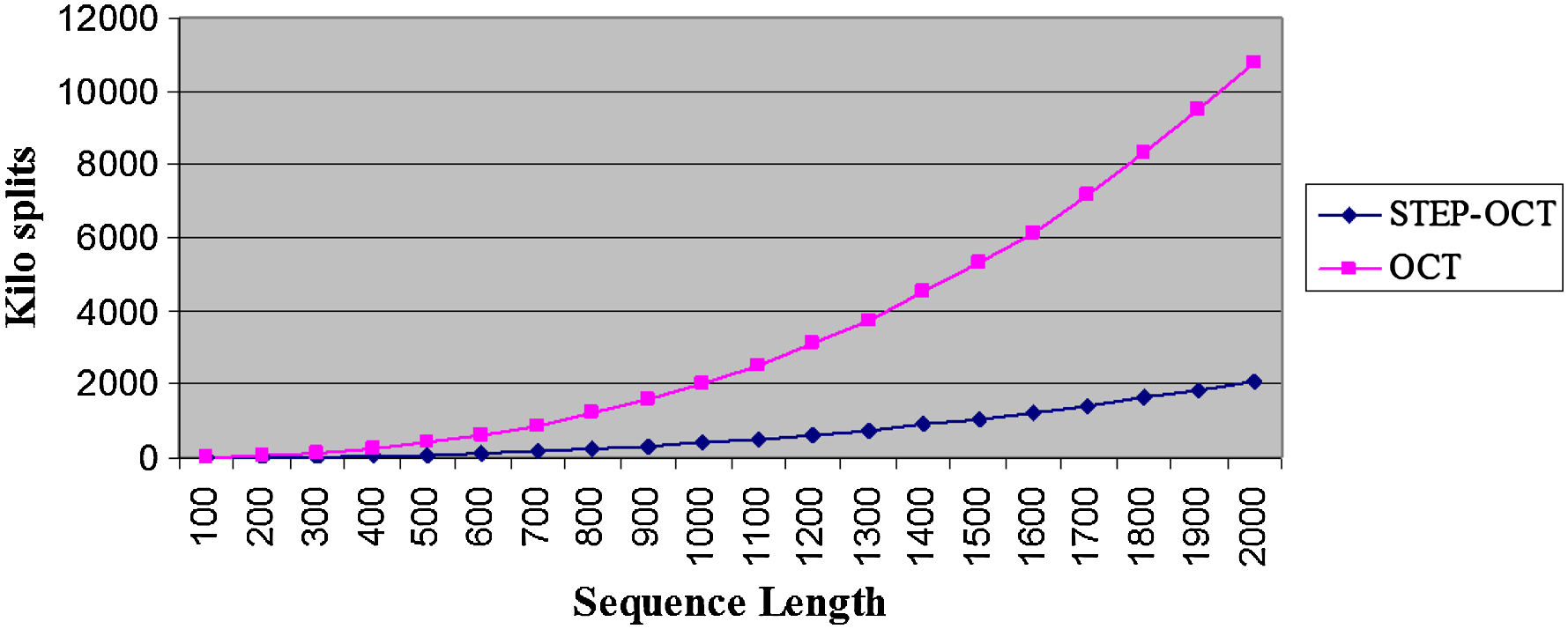
Average examined split points per sequence length. The figure shows only the OCT and STEPOCT curves of Figure 5.
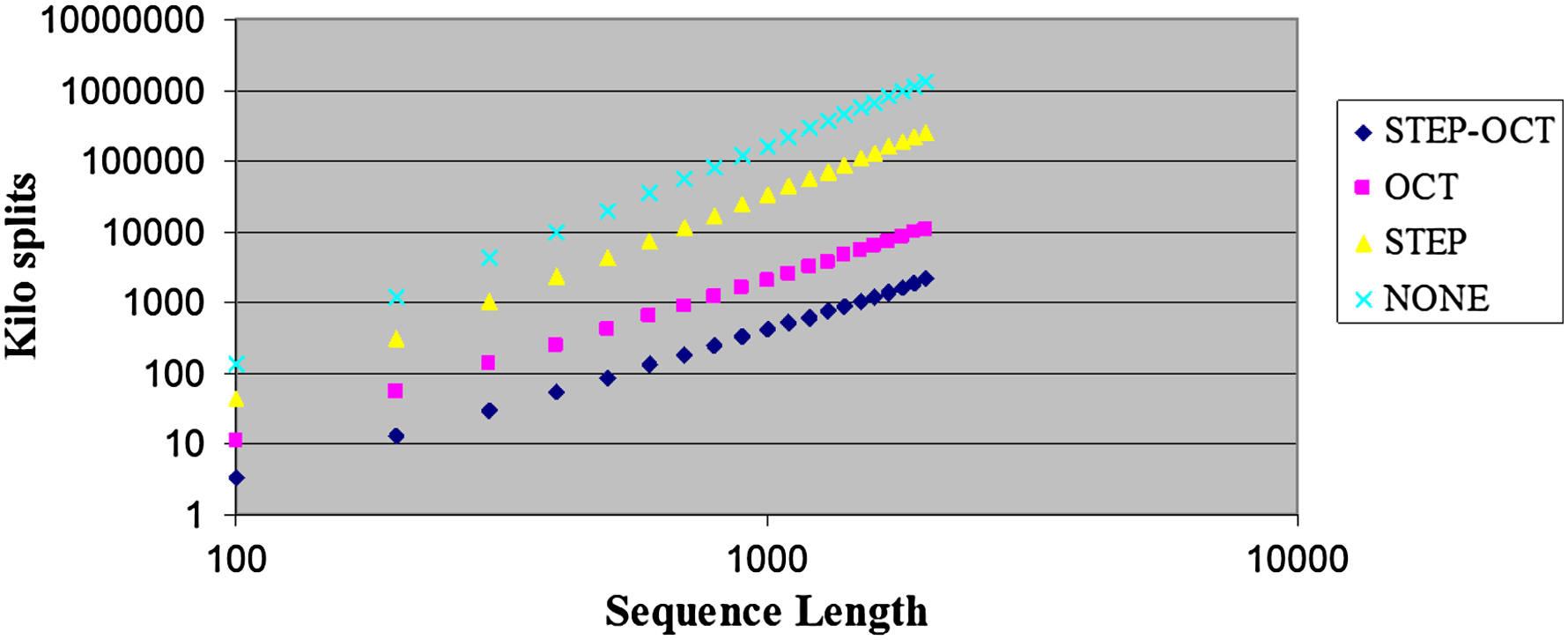
Average examined split points per sequence length. A log-log scale representation of Figure 5.
Footnotes
Acknowledgments
We are grateful to Mirela Andronescu for her kind help in providing information and pointing us to relevant data, and to Holger Hoos and the anonymous reviewers for their helpful comments. The research of S.Z. and M.Z.-U. was supported by ISF grant 478/10.
Disclosure Statement
No competing financial interests exist.
1
Some scoring models also utilize homology information with respect to two or more sequences. Such comparative approaches are beyond the scope of this work.
2
In models whose scores correspond to free energies, the score optimization is traditionally formulated as a minimization problem. This formulation can be easily transformed to the maximization formulation that is used here.
3
In this sense, internal-loop lengths are not restricted here as done in some other models, where arbitrary-length internal-loops are scored with respect to their unpaired bases and terminating base-pairs, and length-depended corrections are added to the scores of relatively “short” loops.
4
This is a common setting in online learning algorithms; for a justification based on an analysis of stochastic-gradient-descent optimization, see Zhang (2004).