
Abstract
Abstract
Protein-protein interactions play an important role in determining the regulatory output of cis regulatory regions. In this work, we revisit the regulatory output functions recorded for the synthetic enhancers that contain binding sites for TetR. We use our thermodynamic model as an analysis tool to infer that two different types of interactions may take place between the TetR molecules. First, a strong mutually exclusive anti-cooperative interaction precludes the synthetic enhancer from being occupied by more than one AT (the aTc bound TetR isoform) protein, and a second weak cooperative interaction exists between the aTc-free TetR isoform (T). Consequently, this work highlights the power of the synthetic enhancer approach as a tool for studying protein-protein interactions via an experimentally verifiable prediction for the general mode of binding of the TetR repressor.
1. Introduction
One approach for studying multiple binding site cassettes is via synthetic biology. Using such a methodology, multiple-binding site cassettes can be decoupled from their ambient regulatory contexts, and as a result can be systematically dissected and analyzed in an independent fashion. Recently, we developed a synthetic regulatory methodology, termed synthetic enhancers (Amit et al., 2011), which can also be used to address these complex questions. Natural enhancers are ubiquitous non-gene coding genomic regions present in all domains of life (Buck et al., 2000), which are capable of integrating the binding of several transcription factors for the purpose of regulating gene expression. We showed that it is possible to build in bacteria new enhancers from “biological parts,” which generate complex regulatory responses to variable inputs. We began with a minimal enhancer architecture that consisted of the NRI/NRII (NtrC/NtrB) two-component system (Magasanik, 1993; Ninfa and Atkinson, 2000) and its associated poised σ54 promoter (Buck et al., 2000; Rappas et al., 2007) in Escherichia coli. To this minimal architecture, we added cassettes of either TetR or TraR binding sites (upstream of the driver or NRI binding sites, and up to 325 bp from the promoter). We used cassettes of one, two, three, and six binding sites, respectively, and with varying binding site spacing. Our experiments showed that synthetic enhancers with either TraR or TetR binding sites repress transcription in values that strongly depend on the number of binding sites on the cassette, and that it was possible to generate output functions that exhibit non-monotonic step-like behavior by systematically titrating the protein level in our cells using ligands (e.g., anhydrous-Tetracycline [aTc]) that either promote or inhibit DNA binding (Amit et al., 2011).
We complemented our experimental approach by developing a modular or nested set of thermodynamic models that reproduced faithfully, although with increasing number of parameters, the experimental regulatory output behavior. Our models captured the main experimental features exhibited by the synthetic enhancers using three simple concepts: the statistics of binding site occupancy, the looping J-factor, and a hypothetical protein-protein interaction between adjacently bound proteins, which can be cooperative or anti-cooperative (see Theory section). However, since we did not pursue the theoretical analysis further, the question remains whether our modeling scheme can generate an additional insight into the nature of TetR binding to multi-binding site cassettes.
2. Methods and Results
Fitting the TetR synthetic cassette data
In order to answer this question and demonstrate the applicability of the synthetic enhancer approach to further characterizing localized protein-protein interactions, we revisit the regulatory output recorded for our first generation of synthetic enhancers made with multiple binding sites spaced at 16 bps for the ubiquitous repressor TetR as enhancer binding protein. TetR is a dimeric protein, which has two relevant structural features: a DNA binding helix-turn-helix domain, and a ligand binding pocket that can bind tetracycline or one of its many homologues with varying affinity (Lederer et al., 1995, 1996; Ramos et al., 2005). As a result, TetR can exist in the cell in three isoforms: the ligand unbound form (T), a TetR dimer bound by a single ligand (AT), and a TetR dimer bound by two ligands (ATA). In vitro studies have shown that only the first two isoforms can bind DNA in a specific fashion (Lederer et al., 1995), with the AT form having its binding affinity reduced by ∼3 orders of magnitude as compared with the ligand-free form (T). In addition, structural studies (Orth et al., 2000) have shown that a bound ligand triggers conformational changes in the protein structure that in turn hinders the DNA binding residues from being able to make contact with the DNA.
In order to gain an insight into the molecular mechanism that underlies the TetR protein-protein interactions that take place on the synthetic enhancer, we utilized two plausible varieties of our thermodynamic model for the regulatory output as an analysis tool (see Theory section). The model versions differ by protein-protein interaction scenarios, which are studied in detail. We do this by exploring the parameter space of the protein-protein interaction portion of our model for solutions that reproduce the major features (i.e., steps, step level, transition slope, etc.) exhibited by the data sets for the synthetic enhancers with two and three TetR binding sites, while simultaneously constraining the search space with experimental observables (for values used in the analysis, see Table 1).
Experimental values for the single occupancy repression level for the two binding site synthetic enhancer (Amit et al., 2011).
Experimental values for the single and double occupancy repression level for the three binding site synthetic enhancer (Amit et al., 2011).
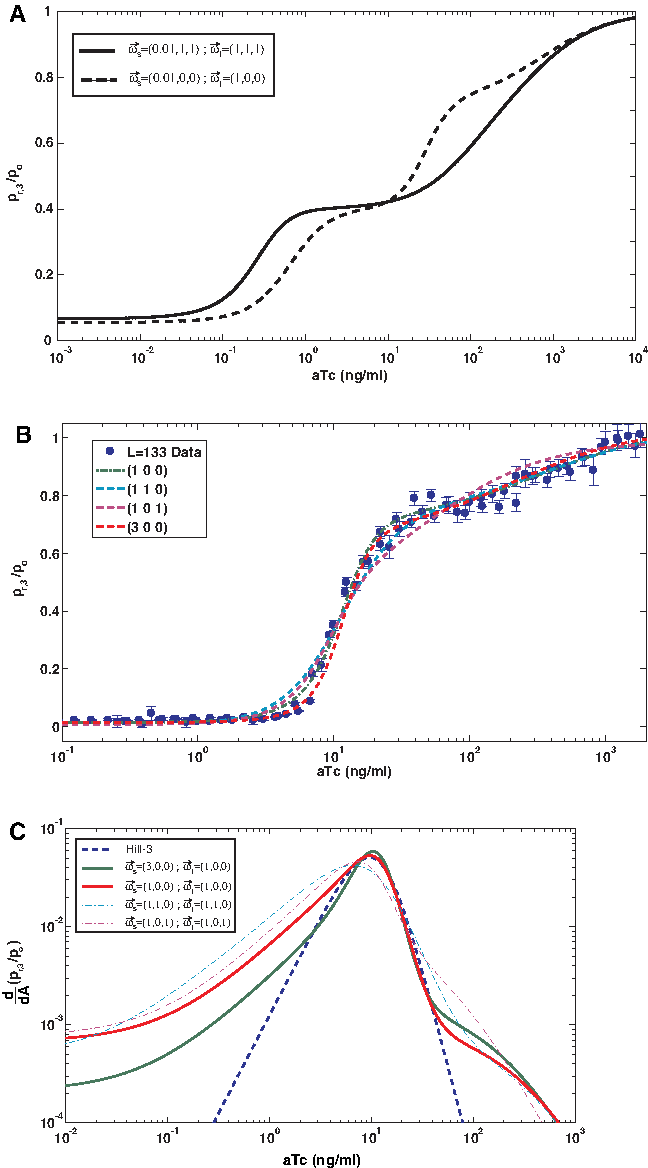
We begin by analyzing the regulatory output function for the synthetic enhancer with two TetR binding sites using the scalar version of our model (Fig. 1A, B). In this case, we model our data with two binding constants for T and AT, and a single or scalar protein-protein interaction parameter that encompasses all types of DNA bound TetR interactions: TetR-TetR (T-T), aTc-TetR-TetR (AT-T), and aTc-TetR-aTc-TetR (AT-AT). In Figure 1A,B, we show the enhancer occupancy probabilities (i.e., probabilities for no occupancy, one binding site occupied, and both binding sites occupied) and regulatory output respectively as a function of ligand (aTc) concentration for two values of the scalar short-range interaction parameter ωs = 1 and 0.001. The panels show that for the case of no interaction (ωs = 1) there is significant overlap between the various occupancy probabilities, which yields a repression level output characterized by a two-state function. However, for a value of ωs < 1, which indicates an anti-cooperative interaction between two bound TetR proteins, the doubly occupied states are destabilized resulting in a shift of its occupancy probability curve to lower aTc values. This, in turn, allows for a wide range of aTc concentrations over which the most likely occupancy state of the cassette is that with only a single TetR bound, which yields a regulatory output function characterized by an intermediate step as exhibited by the data.

Scalar model unable to reproduce 3-Tet step function. The figure shows various output functions using the scalar model.
In order to validate this version of the model, we needed to ensure that a scalar protein-interaction scheme can also reproduce regulatory data recorded for the synthetic enhancer with three TetR binding sites, where in this case we have added a scalar protein interaction parameter for the next-to-nearest neigbor interaction as well. In Figure 1C, we plot the various occupancy probabilities which in a similar fashion to the double occupancy cassette produce no intermediate steps when (ωs = 1,ωl = 1). For the case where an anti-cooperative interaction is inserted into the model (i.e., either ωs < 1 or ωl < 1), the triply-bound state is highly destabilized resulting in a signficantly increased range in aTc concentrations over-which the double occupancy is the most probable occupancy state, while only a small change in the occupancy probability distribution for the single occupancy states is predicted. This, in turn, translates to a prediction that an intermediate step should appear at repression values that closely match that of a double occupancy repression levels measured to be ∼0.4 in our experiments (Table 1). Since this prediction is not supported by the data, which exhibit an intermediate step at much larger values of repression (∼0.7–0.8—commensurate with the single occupancy repression levels), we are forced to conclude that the scalar thermodynamic model fails to describe our data sets in a consistent fashion.
Modeling the triple occupancy state with the vector model
Given this set back, is it possible to generate a model where the single occupancy states are the most probable intermediate states for both the two and three binding site architectures? As was discussed above, TetR can bind DNA in two isoforms (T and AT), allowing us to assume that the binding sites can bind specifically two independent proteins. This implies that we can imagine three independent values for the nearest

Vector model produces correct step function via elimination of states.
Exploring the parameter space for the vector model may be prohibitive, but at this point we are only interested in finding a solution that reproduces the intermediate step at a repression level that matches our experimental observations. In order to show that this model is a natural extension of the scalar model, we plot in Figure 2A the model results with all interaction parameters set to 1, and highlight with dashed lines the various substates that are associated with each bound occupancy state. For instance, for the triple occupancy state there are eight substates reflecting the occupancy statistics of T and AT binding. Namely, a state with three T proteins bound, three degenerate states with two T and a single AT bound, three degenerate states with two AT and a single T bound, and a state with three AT proteins bound.
Since the scalar model is equivalent to the vector model with each of the vector components set to an identical value, we assumed different values for the various protein interaction parameters in order to access a larger space of model solutions. One way of doing this is to assume that the AT form of TetR cannot bind DNA in the vicinity of another bound TetR protein and is completely anti-cooperative, while the T form is ambivalent or non-interactive with other TetR proteins. This implies that any interaction parameter associated with AT is set to 0, while the values for
For completeness, we apply the (1 0 0) model to the case of a synthetic enhancer with two TetR binding sites. As for the case shown in Figure 1B for the scalar model, we obtain an intermediate level at the single occupancy state. The consistency of our model function with both the experimental output functions generated for the synthetic enhancer with two and three TetR binding sites allows us to conclude that the vector model in some form provides an adequate set of solutions to all data sets.
Convergence and preferred form of the vector model
Unlike the scalar model, which required a simple mapping of a 2-dimensional phase space for a single short and long-range interaction parameters to determine the range of possible solutions, the vector model is a six-dimensional phase space with three short and long range parameters respectively. Fortunately, the complexity of our search space can be reduced by noticing (Fig. 3A) that values for either
Exploring the vector model solution space. Optimizing the fit to all data sets using the vector model.
To simplify our analysis further, we choose to implement an “integer” vector model to explore in a discrete fashion the range of possible outcomes. Based on the results above, we can constrain this version of the model by demanding that the AT and T interaction parameters will be radically different to ensure that the behavior observed in the experiments can be obtained as follows:
This implies that in general we will only consider combinations of the following classes of interaction parameter values for the short and long range interactions, respectively:
With n ≥ 1 for all cases.
One can compare the different scenarios by examining the goodness of fit with respect to the data (Fig. 3B). However, the solutions seem to cluster with reasonably good fits. A more instructive method to determine which scenario corresponds to the best model is to examine the derivative of the model step function with respect to aTc. By doing so, we can ask which vector model reproduces one of the more compelling features that we previously reported Amit et al. (2011). Namely, that the transition between the strongly repressed state and the first intermediate is characterized empirically by a Hill function of order three for the synthetic enhancer with three TetR binding sites.
Plotting the derivative of each model repression level function as well as the derivative for a Hill function of order three (Fig. 3B), we notice that the (1 0 1) and (1 1 0) models clearly do not reproduce the steepness of the transition region well. This can also be noticed, upon closer inspection of the actual fit to the data (Fig. 3B). In addition, even though the (1 0 0) model seems to adequately reproduce the transition steepness, a model with a slightly cooperative T-T interaction (i.e., (3 0 0)) clearly matches the transition better. Thus, it would seem that the best molecular interaction model for TetR is characterized by a slight cooperativity between adjacently bound TetR proteins, and total anti-cooperativity between atc-TetR molecules and nearby bound proteins that leads to the elimination of most of the double and triple occupancy states.
3. Discussion
In this work, we demonstrated the ability to differentiate between different types of thermodynamic models for synthetic enhancer regulatory output based purely on experimental data. This, in turn, provides an additional insight into the underlying molecular mechanism that guides the binding of TetR to DNA. We first determined that a model version that only posits one type of protein-protein interaction is inconsistent with our data. Instead, we focused on three different types of protein-protein interactions that may take place on an enhancer with TetR as enhancer binding protein. We inferred that ligand-free form (T-T) interactions may be slightly cooperative, but that the ligand-bound AT-T and AT-AT interactions are mutually exclusive or totally anti-cooperative. This means that enhancer occupancy states with more than one protein bound, when one of those proteins is AT are highly unlikely and unstable.
The main conclusion from this analysis appears to be that quantitative data, when analyzed carefully with a thermodynamic model can lead to qualitative insights into the molecular basis of the protein-DNA interactions. In this case, due to the weak cooperative interaction inferred for two adjacently bound TetR molecules at 16 bp spacings, it is tempting to speculate that at the 10 bp spacing associated with the naturally occurring tandem of TetR binding sites on the Tn10 tet operator cassettes in closer proximity Hillen et al. (1984); Hillen and Berens (1994) a larger cooperative interaction will be observed. Therefore, the dual cooperative/anti-cooperative interaction inferred for TetR-DNA binding may have an important regulatory role.
What is the origin of the cooperativity and anti-cooperativity interactions inferred from our model? While many different structural scenarios can be envisioned (e.g. inhibition of DNA wrapping due to the close proximity of binding sites Levandoski et al. (1996); Tsodikov et al. (1999), cumulative localized deformation of DNA due to several binding sites Wang et al. (2005), etc.) our model and data do not provide an additional insight into this question. Therefore, while our data and model can be considered quantitative for most purposes, our inability to draw any further structural conclusions also point to the short-comings of our theoretical and experimental approach.
Finally, it is worth noting that our modeling scheme is still highly qualitative, and corresponds to one possible interpretation of the data presented in Amit et al. (2011). It is certainly possible that other explanations or models can lead to a consistent interpretation of this data as well. However, the experimental scheme presented in Amit et al. (2011) can provide a high-throughput platform that combined with our modeling scheme and additional structural data (i.e. crystallographic, cryo-EM methods, and other in vitro techniques) will be able to eventually generate a quantitative understanding of protein-protein interactions, which at this point is a moniker for many possible different structural mechanisms that may manifest themselves in our experimental scheme. Since many regulatory sequences contain several binding sites clustered in close proximity for one or more proteins, an understanding of all-types of protein-protein interactions at the structural level is crucial for a full decipherment of the regulatory code.
4. Theory
Enhancer regulatory output model: the TetR case
For a thorough model description and key definitions, see Amit et al. (2011). In short, we previously devised a thermodynamic set of models that posit sets of states and weights, which define the probability that an m-binding site synthetic bacterial enhancer bound by n transcription factors would loop and initiate expression. In general, for each enhancer occupancy we defined two sets of states: looped and transcriptionally active
unlooped and transcriptionally inactive,
The looped states are described by a sum of possible occupancy configurations, where each configuration is represented by product of several quantities: a protein binding term
Notice, that for the simpler case, where only one protein-type can bind a binding site, we have 2 times 2m number of states. Namely, 2m looped and 2m unlooped states.
For the case of TetR, where there are two isoforms of the protein that can bind each binding site, we have 3m looped and unlooped states respectively, given the fact that each site can be either occupied with isoform 1, isoform 2, or not occupied at all. As a result, the equations for the looped and unlooped states become:
and the partition function is now given by:
where T and AT are the number of free TetR and aTc-TetR molecules in the cell and can be related to the ligand concentration by simple expressions derived in Amit et al. (2011). KTD and KATD are their respective binding constant, and note that for the special case of n = 2, ωl is always set to one.
States and weights thermodynamic model for synthetic enhancer occupancy by TetR
Throughout this article, we plot two types of figures. The first show regulatory output functions for the two and three binding site synthetic enhancer derived from a model developed and described in detail in Amit et al. (2011), and briefly summarized above. The second set of plots depict the different enhancer occupancy probability distributions for TetR (T) and its aTc occupied isoform aTc-TetR (AT). The purpose of these plots is to show where steps in the regulatory output should emerge given the values of the protein interaction parameters, and their effect on the shape of the occupancy states probability distributions.
In order to derive an expression for the occupancy probabilities from the model described above, we first note that the partition function for occupancy probability consists of only TetR occupancy states and does not involve looping. This implies that the occupancy partition function for an m-TetR binding site synthetic enhancer becomes:
As an example, consider the case of the synthetic enhancer with two TetR binding sites. In this case there are nine occupancy states: one state where the enhancer is not occupied, four states where the enhancer is occupied with either T or AT at one of the two binding sites, and four states where the enhancer is occupied with two proteins in the following possible configurations: T/T, AT/T, T/AT, and AT/AT for the proximal and distal sites respectively. Using this information, eqn (8) becomes:
which leads to the following expressions for the different occupancy probability distributions:
for the no occupancy, single occupancy, and double occupancy probabilities respectively.
Expanding this model to the case where the synthetic enhancer contains three binding sites is a matter of accounting for all 27 occupancy states. In this case, there is a single unoccupied state as before, 6 states with a single AT or T bound, 12 states with two sites bound in some configuration, and 8 states with all three binding sites occupied in some configuration. This leads to the following partition function:
which in turn leads to the following probabilities:
whose curves as a function of aTc concentration are plotted in the figures of the text. In addition, for all the cases in the text where the various short range and long range protein-protein interaction parameters (ωs and ωl) are set to identical values, which do not discriminate between protein isoforms, eqn. (12) and (10) reduce to the scalar model versions of our model whose output functions and probability plots are depicted in Figure 1.
Footnotes
Acknowledgments
I would like to thank Rob Phillips and Hernan G. Garcia for early discussion of the ideas that appear in this article.
Disclosure Statement
No competing financial interests exist.