
Abstract
Abstract
Sequence alignment is an important tool for identifying regions of similarities among proteins and for, thus, establishing functional and structural relationships between different proteins. Here, alignments of transmembrane domains (TMDs) of viral channel forming proteins with host ion channels and toxins are evaluated. The following representatives of polytopic viral channel proteins are chosen: (i) p7 of HCV and 2B of Polio virus (two TMDs) and (ii) 3a of SARS-CoV (three TMDs). Using ClustalW2, each of the TMDs of the viral channels is aligned, and the overlap is mapped onto structural models of the host channels and toxins focusing on the pore-lining TMDs. The analysis reveals that p7 and 2B TMDs align with the pore-facing TMD of MscL, and 3a-TMDs align with those of ligand-gated ion channels. Possible implications concerning the mechanism of function of the viral proteins are discussed.
Introduction
Providing structural information for VCPs is thought to be vital to understanding their role in viral replication. In addition, sequence similarity with other channel proteins implies close functional relationships (Anfinsen et al., 1961; Chothia and Lesk, 1986), despite exceptions (Blake and Cohen, 2001). Sequence–structure–function relationships can be based on similarities on the level of the fold, which are called globular similarities, or on the level of motifs, the so-called local similarities (Hvidsten et al., 2009; Thornton et al., 2000). It is assumed that the limited number of 20 natural amino acids restricts sequence diversity for mechanical motifs such as hinges or pivot points. Besides, the environment in which channel proteins operate (i.e., a lipid membrane) further restricts the number of amino acids that can be used, making high sequence diversity very unlikely (Oberai et al., 2009).
Bioinformatics and computational structural modeling are important tools for obtaining structural and functional information when experimental data are lacking (Lee et al., 2007). In this work, we examine sequence similarities for several VCPs: p7 from Hepatitis C virus, 2B from Polio virus, and 3a from SARS-CoV. For each of these viral proteins, high-resolution three-dimensional structure data are not available to date.
The VCPs (Fischer and Sansom, 2002; Fischer et al., 2011; Gonzales and Carrasco, 2003) are capable of changing the permeability of ions (Campanella et al., 2004; Griffin et al., 2003; Lu et al., 2006; Pavlovic et al., 2003; Premkumar et al., 2004) and small molecules (2B) (Aldabe et al., 1996; Doedens and Kirkegaard, 1995; van Kuppeveld et al., 1997a) across a lipid membrane. Their biological roles are not yet fully explored (Fischer and Hsu, 2011; Fischer and Sansom, 2002; Gonzales and Carrasco, 2003).
Protein p7 is a 6-to-7-kilodalton polytopic membrane protein of 63 amino acids (Elbers et al., 1996; Lin et al., 1994). It is expressed as part of a large polyprotein and is preceded by the structural protein E2 and succeeded by the nonstructural protein NS2. Whether p7 belongs to either of these sides is not yet fully elucidated. The protein is essential for the virus inasmuch as it locates NS2 to the site of viral replication and particle assembly (Tedbury et al., 2011).
Besides this function, it has been concluded from in vitro studies that p7 also acts as a VCP (Griffin et al., 2003; Pavlovic et al., 2003; Premkumar et al., 2004; Sakai et al., 2003). Structural details emerging from nuclear magnetic resonance (NMR) spectroscopic experiments reveal an antiparallel alignment of its two transmembrane domains (TMDs) in membrane systems (Cook and Opella, 2011; Montserret et al., 2010). From electron microscopy, it is deduced that the protein assembles most likely into a hexameric bundle (Griffin et al., 2003; Luik et al., 2009; Patargias et al., 2006), although a heptameric form has been recorded as well (Clarke et al., 2006). A computational model suggests that a histidine residue is pointing into the putative pore (Patargias et al., 2006), a suggestion that is supported by channel recordings in the presence of cuppor ions (Chew et al., 2009).
2B from Polio virus is a 97 amino acid VCP with two TMDs (van Kuppeveld et al., 1995; van Kuppeveld et al., 1997b), which most likely forms tetrameric assemblies (Agirre et al., 2002; Cuconati et al., 1998; de Jong et al., 2002). It is proposed that 2B retains in the endoplasmic reticulum and Golgi apparatus, where its role is to increase membrane permeability to Ca2+ and modulate Ca2+ homeostasis (Campanella et al., 2004; de Jong et al., 2006). In addition, permeability to small molecular weight compounds (Aldabe et al., 1996; Doedens and Kirkegaard 1995; van Kuppeveld et al., 1997a) and monovalent anions (Xie et al., 2011) has also been reported. Detailed structural information has not yet emerged, except for a computational model based on molecular dynamics simulation and secondary structure predictions (Patargias et al., 2009). In this study, it is proposed that both TMDs line the pore, and lysine residues of TMD1 contribute to the lumen of the pore.
Expression of 3a from SARS-CoV in Xenopus oocytes in combination with whole cell channel recordings has shown that this protein induces channel activity (Lu et al., 2006). It has been suggested that this function is important for viral release (Lu et al., 2006) and membrane rearrangements (Freundt et al., 2010). Protein 3a is reported to be a three TMD containing 274 amino acids membrane protein, which is found to be a tetramer with two monomers being covalently linked via disulphide bridges (Lu et al., 2006). Together with other proteins from SARS-CoV, 3a is regarded as an accessory protein (Narayanan et al., 2008), albeit deletion studies have not yet been reported to date. Structural information is currently only available through computational models (Hsu and Fischer, 2011; Krüger and Fischer, 2009) It is proposed that, depending on the modeling protocol, TMD2 (Hsu and Fischer, 2011) or TMD3 (Krüger and Fischer, 2009) mantle the pore. In addition, residues like histidines (Hsu and Fischer, 2011) or tyrosines (Krüger and Fischer, 2009) are suggested to be pore lining.
With the sequence–structure–function relationship in mind, potential mechanistic details of the viral proteins are assessed by sequence comparison with those channels and pores for which structural information is available (see also Fischer and Hsu, 2011). The focus is put on the TMDs—pore-lining residues, in particular—of the viral and host channels, as well as toxins. The idea is to derive information on the mechanism of function of the VCPs based on sequence similarity. The results should also elucidate potential routes for drug discovery based on the idea that with similar sequence and fold, similar drugs should be effective.
Materials and Methods
The respective TMDs of the viral channel proteins have been chosen as follows.
p7 from HCV (P27958, isolate H; www.uniprot.org) (Pavlovic et al., 2003): p713–32, LAGTHGLVS FLVFFCFAWYL (TMD1), p739–57, GAVYALYGMWPLLLLLLAL (TMD2) (Patargias et al., 2006); 2B from Polio virus (van Kuppeveld et al., 1997b): 2B1–18, TITEKLLKNLIKIISSLV (TMD1), 2B27–46, TTTVLATLALLGCDASPWQW (TMD2) (Patargias et al., 2009); 3a from SARS-CoV (GenBank: ABA02268.1, www.ncbi.nlm.nih.gov) (Li et al., 2003): 3a35–55, IPLQASLPFGWLVIGVAFLAV (TMD1), 3a79–99, FICNLLLLFVTIYSHLLLVAA (TMD2), 3A105–125, FLYLYALIYFLQCINACRIIM (TMD3) (Krüger and Fischer, 2009).
These channel proteins were aligned against Vpu from HIV-1 (GenBank: AAB59750.1, www.ncbi.nlm.nih.gov); Cytolysin A (ClyA, toxin; protein data bank code: 2WCD) (Mueller et al., 2009); α-hemolysin from Staphylococcus aureus (7AHL) (Song et al., 1996); acid-sensitive potassium channel protein (TASK, UniProtKB/Swiss-Prot O14649, www.uniprot.org) (Hsu et al., 2004); K+ channel from Streptomyces lividans (KcsA, 1BL8) (Doyle et al., 1998); nicotinic acetylcholine receptor (nAChR, 2BG9) (Unwin, 2005); α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-sensitive homotetrameric rat glutamate A2 receptor (GluA2, 3KG2) (Sobolevsky et al., 2009); prokaryotic pentameric ligand-gated ion channel (ELIC, 2VL0) (Hilf and Dutzler, 2008); pentameric formate channel (pFC, 3KLY) (Waight et al., 2010); large-conductance mechanosensitive channel (MscL, 2OAR) (Chang et al., 1998); and bone marrow stromal antigen 2 (BST-2, UniProtKB/Swiss-Prot Q10589, www.uniprot.org) (Ishikawa et al., 1995).
Proteins have been aligned in pairs using ClustalW2 (www.ebi.ac.uk). The following settings were used: gap penalty = x, extended gap penalty = x, delay divergence = x, gap distance = x. The default settings of ClustalW2 were used. These are given as follows: DNA Gap Open Penalty = 15.0, DNA Gap Extension Penalty = 6.66, DNA Matrix = Identity, Protein Gap Open Penalty = 10.0, Protein Gap Extension Penalty = 0.2, Protein matrix = Gonnet, Protein/DNA ENDGAP = −1, Protein/DNA GAPDIST = 4.
Results
Mapping to pore-facing residues observed
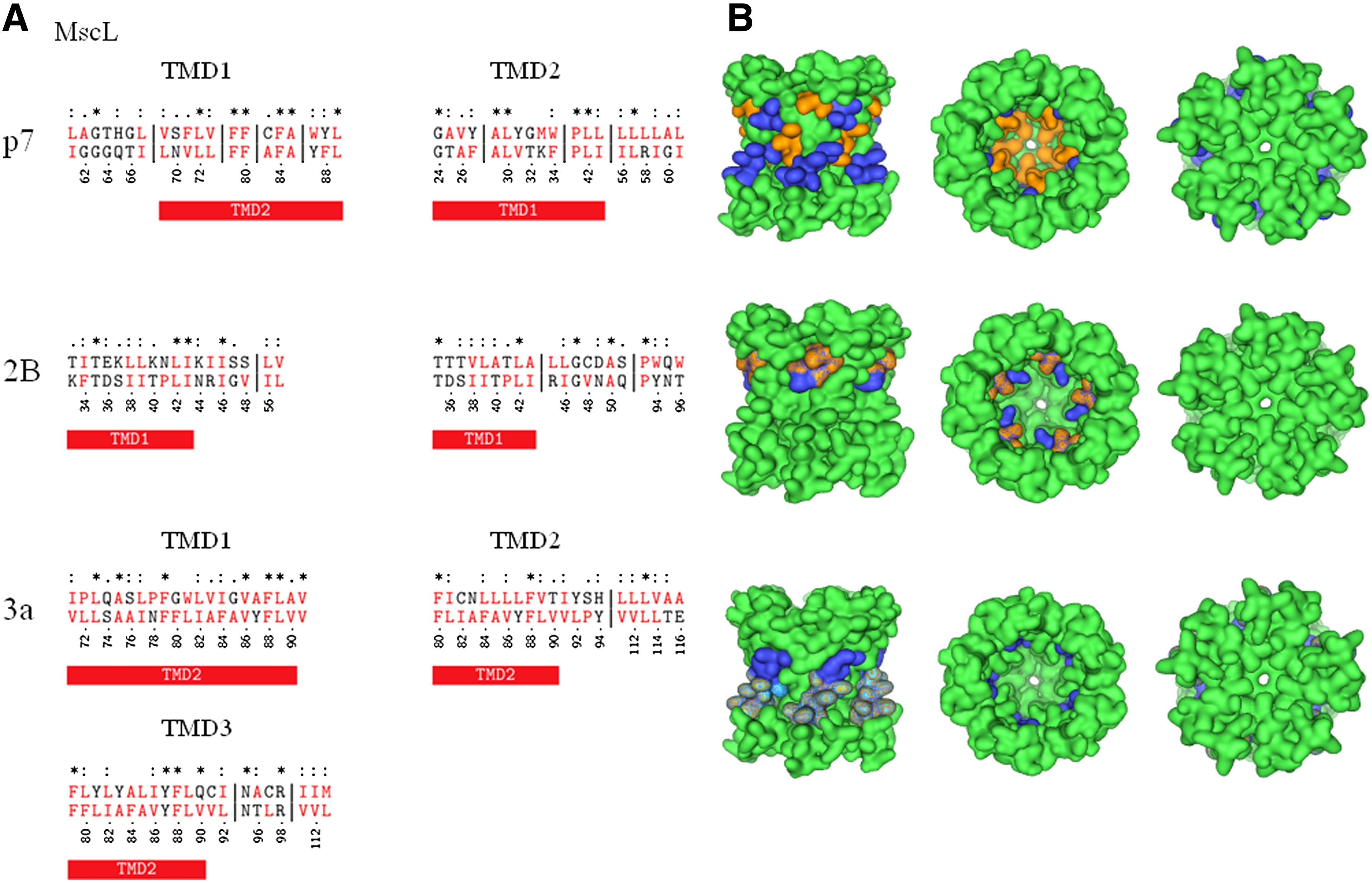
Alignment with MscL
Alignment of the entire length of p7 matches the sequence of the cytoplasmic part of MscL with about nine gaps (data not shown). Truncating the cytoplasmic part of MscL results in an overlap of the C terminal side of p7-TMD1 with MscL-TMD2 (Fig. 1, upper left panel; Table 1A,B). The alignment results in six identical residues (2 × F, 2 × L, A) and three gaps of 5, 2, and 1 residues in MscL-TMD2. In both sequences, a pattern of aromatic amino acids W-30/Y-31 (p7-TMD1) and Y-87/F-88 (MscL-TMD2) mark the end of the helices. The N terminal side of p7-TMD2 aligns with MscL-TMD1 (Fig. 1 upper left panel; Table 1A,B) with five identical residues and two gaps (1 and 6 residues). Besides an overlapping sequence of hydrophobic residues, two prolins in either chain match (P-49 (p7-TMD2) and P-41 (MscL-TMD1). Aromatic amino acids (W-48 (p7) and F-34 (MscL) match as well. Visualization of the overlap reveals that p7-TMD1 is matching the outer helix, and p7-TMD2 is matching the inner pore-lining helix of MscL (Fig. 1, upper right panel).
(
Entry Code in the protein data bank (www.rcbs.org) is given in brackets. Identical aromatic amino acids are listed. The helices of the toxin and the nonviral channel proteins are named according to their respective sequence number in the literature.
“Aa” = amino acids; “*” = identical residues; “:” = conserved substitution; “.” = semi-conserved substitution; “-” = no substitution; “TMD” = transmembrane domain.
Both TMDs of 2B align each with their N terminal side to MscL-TMD1 (Fig. 1 middle left panel; Supplementary Table 1A,B; Supplementary Material is available online at www.liebertonline.com/cmb). Identical residues found are 3 and 2 for 2B-TMD1 and 2B-TMD2, respectively, without any gaps. Both 2B-TMDs overlap with the C terminal side of MscL-TMD1. For both 2B TMDs, the matching residues are T, L, and I. Both 2B TMDs show a hydrophilic motif of TEK (2B-TMD1) and TTT (2B-TMD2) that match a similar triplet of TDS of MscL-TMD1, which is located in the middle of the MscL TMD1. Mapping the overlap onto the crystal structure of MscL reveals residues from both of the 2B-TMDs facing the lumen of the MscL channel (Fig. 1, middle right panel).
3a-TMD1 shows an alignment of its entire length with MscL-TMD2 (Fig. 1 lower left panel; Supplementary Table 2A). There are seven identical residues (2 × L, A, V, and F) and both sequences show no gaps. Both 3a-TMD2 and 3a-TMD3 align their N terminal sides with the C terminal side of MscL-TMD2 (Fig. 1; Supplementary Table 2B,C). There are no gaps, and there are 2 (2 × F) and 3 (2 × F, Y) identical residue alignments for 3a-TMD2 and 3a-TMD3, respectively. With MscL-TMD2 being partially aligned to the pore with its C terminal side, 3a-TMD1 only marginally matches the pore alignment (Fig. 1, lower right panel).
Alignment with nAChR
Sequence alignment of p7-TMD1 with nAChR results in no alignment with any of the TMDs of the subunits of the receptor. The hydrophobic stretch of p7-TMD1 is, rather, placed within the β-sheet and loop regions of the chains (data not shown). For B, C, and E chains of nAChR, p7-TMD2 aligns with the M1 domain of chain B and M4 domain of chains C and E (Fig. 2A; Table 1A,B). In the case of the C-chain, there is considerable overlap of p7-TMD2 with the N terminal side of the M4 domain, which leaves nine residues of p7-TMD2 matching two identical residues (P, L), separated by a short gap of four residues. For the E chain, the match extends over the entire length of the M4 region, resulting in nine identical matches (4 × L, 2 × G, W) and three gaps of 3, 7, and 4 residues. Highlighting the overlapping residues onto the structure of the nAChR leaves parts of p7-TMD2 matching with the domain at the protein lipid interface (Fig. 2A).
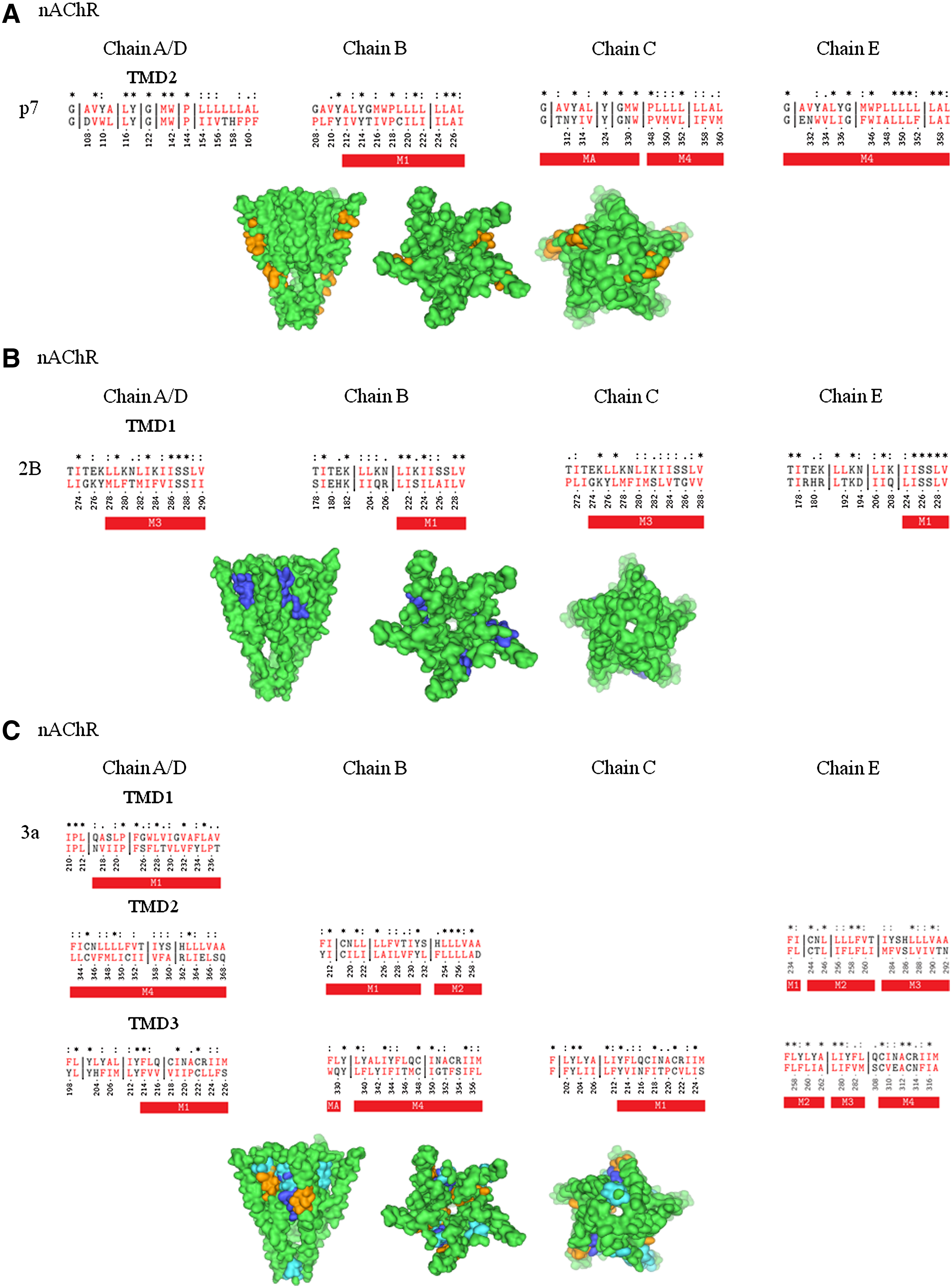
Sequence alignment of p7 (HCV) (
2B-TMD1 matches M1 (chain B and E) and M3 domains (chain A/D and C) of the nAChR without any gaps (Fig. 2B; Supplementary Table 1A,B). 2B-TMD2 is placed into the extramembrane part of the receptor, independent of the chain analyzed. The C terminal side of 2B-TMD1 aligns to short segments of the N terminal side of the M1 domains, with six residues of chain E, including five identical residues and a double S motif, and to nine residues of chain B, including five identical residues, mainly hydrophobic. Overlap with M3 of nAChR covers a larger range of 13 residues of 2B-TMD1 with M3 of chain A/D, including 5 identical residues (double S motif and hydrophobic residues) and 11 amino acids of chain C, including 3 identical residues (hydrophobic and K). With this alignment, stretches of 2B-TMD1 are similar to stretches of the nAChR, which are located at the protein–lipid interface (Fig. 2B, middle panel).
All three 3a-TMDs align with TMDs of chain A/D of nAChR (Fig. 2C; Supplementary Table 2A–C). The number of overlapping residues with chains of the nAChR increases from 3a-TMD1 (Chain A/D) and 3a-TMD2 (Chain A/D and B) to 3a-TMD3, with overlap in all chains. The overlap with individual nAChR-TMDs is in most cases interrupted by gaps. Overlap with no gaps are detected for 3a-TMD2 and parts of M2 (Chain B) and M3 (Chain E), as well as for 3a-TMD3 and parts of M2 and M4 of chain E. None of these gap-free overlaps exceed 10 residues. Identical residues are mainly hydrophobic. Having a sole overlap with the N terminal side of chains B and C, 3a hardly matches with the pore-lining helix M2 (Fig. 2C).
Alignment with ELIC
Only p7-TMD2 aligns with parts of pore-lining α2 (7 residues, 3 identical (2 × Y), one gap with 2 residues) and with α3 (9 residues with one larger gap of 19 residues) of ELIC (Fig. 3, upper panel; Table 1B). Alignment of p7-TMD1 screens a β-sheet region of the ion channel (data not shown).
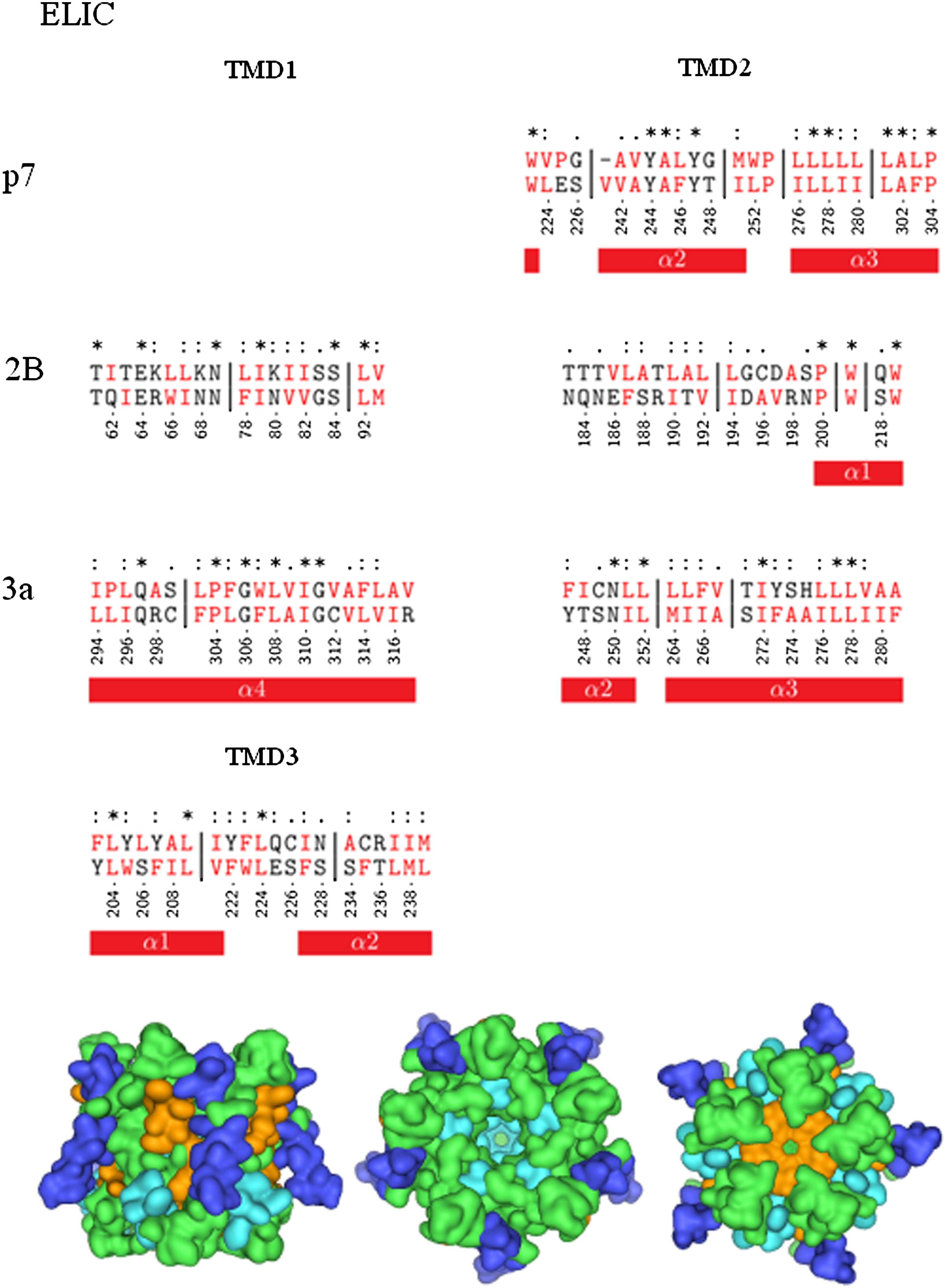
Sequence alignment of p7 (HCV), 2B (Polio virus), and 3a (SARS-CoV) with prokaryotic pentameric ligand-gated ion channel (ELIC, 2VL0). The symbols are defined as follows: “*” = identical residues; “:” = conserved substitution; “.” = semi-conserved substitution; “-” = no substitution. Vertical lines indicate gaps. Hydrophobic residues are shown in red. The red bars indicate helical TMDs of the proteins. The helices of ELIC are named according to their respective notation in the literature. Bottom: Mapping of the aligned sequences of 3a onto the crystal structure (green) of ELIC. The color code for mapping is as follows: Alignments according to viral TMD1s are shown in blue, those according to TMD2s in orange, and those according to TMD3s of 3a in cyan.
There is no alignment of 2B-TMD1 with any TMD of the channel for 2B. 2B-TMD2 only aligns with its two tryptophans with α1 (Fig. 3, middle panel; Supplementary Table 1B).
All three 3a-TMDs align with TMDs of ELIC: 3a-TMD1 with α4 (21 residues, 1 gap of 3 residues), 3a-TMD2 with α2 (5 residues), and α3 (15 residues, 1 gap of 3 residues), as well as 3a-TMD3 with α1 (7 residues) and α2 (8 residues) (Fig. 3, lower panel; Supplementary Table 2A–C). Identical residues include mostly hydrophobic residues and a proline for 3a-TMD3 with α4. Visualization indicates the alignment of both 3a-TMDs two and three with the pore helices of ELIC even more than for nAChR (Fig. 3, lower panel).
Borderline mapping
Alignment with KcsA
Alignment of full-length p7 with KcsA results in an overlap of p7-TMD1 with the N terminal part of KcsA (data not show). In the same alignment, p7-TMD1 shows overlap with the region of the inner helix. A separate alignment of p7-TMD1 and p7-TMD2 places each of the domains within the region of the inner helix of KcsA (KcsA: 88–111; Table 1A,B; Supplementary Fig. 1A, upper panel). P7-TMD1 shows two gaps of eight and one residues and nine identical residues. P7-TMD2 shows one gap with one residue but no identical residues. The hydrophobic stretch matches perfectly well with the hydrophobic stretch LVAVVVMVA of KscA. An optical illustration of the overlap on the crystal structure of KcsA reveals that the overlap of p7-TMD1 and p7-TMD2 with the inner helix is visible from outside the pore for p7-TMD1 (Supplementary Fig. 1A, upper panel).
Separate alignment of the two 2B-TMDs with KcsA reveals an alignment of only the 2B-TMD2. An uninterrupted stretch of 12 residues matches with the outer helix of KscA, including five identical residue matches (Supplementary Fig. 1A, middle panel). Only two residues, 2B W-44 and 2B Q-45, match with the pore helix of KcsA (Supplementary Fig. 1A, middle panel; Supplementary Table 1B).
Alignment of 3a-TM1 and 3a-TM2 reveals an overlap of 10 and 11 consecutive residues of their C terminal sides with the N terminal side of the outer helix of KcsA (Supplementary Fig. 1A, lower panel; Supplementary Table 2A,B). 3a-TM3 shows alignment of 16 consecutive residues with the inner helix of KcsA (Supplementary Fig. 1A, lower panel; Supplementary Table 2C). The N terminal sides of 3a-TM1 and 3a-TMD2 are placed along residues of the cytoplasmic part of KscA, whilst for 3a-TM3, overlap with the extracellular part is observed. Visualization of the overlap reveals that 3a-TMD1 and 3a-TMD2 are seen from the outside and only 3a-TMD3 would match with the inside of the pore. With parts of p7 (TMD1) and 3a (TMD-3) aligning with the inner helix, KcsA reveals some similarity with these two channels.
Alignment with TASK
The p7 protein only aligns with a short N terminal segment of its TMD2 (Table 1B; Supplementary Fig. 1B, upper panel). The 2B protein aligns with both of its TMDs over a much longer stretch of the TM3 domain of TASK (Supplementary Fig. 1B, middle panel; Supplementary Table 1A,B). All three TMDs of 3a match with the TMDs of TASK. 3a-TMD1 aligns with TM4, 3a-TMD2 with TM1, and 3a-TMD3 with parts of TM2 and TM3. (Supplementary Fig. 1B, lower panel; Supplementary Table 2A–C).
Weak or no mapping to pore-facing residues observed
GluA2
Alignment of full-length p7 sequence with GluA2 identifies an overlap of p7-TMD1 with residues 481–519 of the extracellular domain of GluA2 (residues 1–522; data not shown). The individual p7-TMDs both align with M4, one of the TMDs of GluA2 (Supplementary Fig. 1C, upper panel; Table 1A,B). The alignment is almost with the entire stretch of the TMDs of the two proteins. Both p7-TMDs show only two gaps. There are six identical residues in each of the p7-TMDs. M4 of GluA2 is located on the outside of the GluA2-TMD facing the lipid membrane (Supplementary Fig. 1C, upper panel).
2B-TMD1 aligns with M3 and 2B-TMD2 with M2 of GluA2 (Supplementary Fig. 1C, middle panel, Supplementary Table 1). 2B-TMD1 overlaps with the entire stretch of M3, whilst just eight residues of 2B-TMD2 match with M2. The two tryptophans of 2B-TMD (W-48 and W-50) match with M3. This generates a picture of 2B-TMD1 similar to the one entry side of the GluA2 pore and 2B-TMD2 on the other entry side (Supplementary Fig. 1C, middle panel). Within the pore, 2B-TMD2 dominates due to its alignment with M2, which exposes much of its sequence to the lumen of the pore.
In the case of 3a, all 3a-TMDs align almost entirely with M1 of GluA2 (Supplementary Fig. 1C, lower panel; Supplementary Table 2). 3a-TMD1 and 3a-TMD3 both show some overlap with the pre-M1 helix of GluA2. For all 3a-TMDs, three residues are found to be identical (Supplementary Table 2 A,C). Visualization of the results shows that M1 is not forming the lumen but the sheathing of the TM section of GluA2 (Supplementary Fig. 1C, lower panel).
Alignment with pFC
Alignment of p7 with pFC results in considerable alignment of p7-TMD1 with TM6 of pFC and p7-TMD2 with TM1 (7 residues) and TM2a (12 residues) of pFC (Table 1; Supplementary Fig. 1D, upper panel). None of the overlaps reveal a contribution to the pore lining part of the channel (Supplementary Fig. 1D, upper panel).
2B-TMD1 aligns with TM6 of pFC with 18 residues whilst 2B-TMD2 and TM2a align with 12 residues (Supplementary Fig. 1D, middle trace; Supplementary Table 1). Visualization indicates that overlap of TMD2 with TM2a places some of the residues of 2B into the lumen of pFC (Supplementary Fig. 1D, middle panel).
With its TMDs, 3a shows alignment with TM5b and three residues of TM6 for TMD1, for TMD2 with TM2a and TM2b, and TMD3 with TM3 and TM4 (six residues) of pFC (Supplementary Fig. 1D, lower panel; Supplementary Table 2). None of these overlaps are with pore-lining residues (Supplementary Fig. 1D, lower panel).
Alignment with toxins
Alignment with ClyA
An alignment of the two p7-TMDs separately, with full-length ClyA, leaves p7-TMD1 highly scattered (five gaps) along residues 50 to 100 of ClyA (data not shown). The second TMD, p7-TMD2, is aligned along αB and αC of ClyA (Fig. 1E, upper panel; Table 1). Visualization of the alignments of p7-TMD2 is with the extramembrane part of ClyA (Supplementary Fig. 1E, upper panel). Only if full-length p7 is aligned with ClyA, or with a truncated part of ClyA (its first 137 amino acids), p7-TMD1 aligns with αA of ClyA, which is part of the TMD of this protein (see also Fischer and Hsu, 2011). Focusing on the alignment of the individual TMDs of the viral proteins, it is suggested that none of the TMDs of p7 show a reliable alignment with any of the TMDs of ClyA (Supplementary Fig. 1E, upper panel).
Alignment of 2B and ClyA matches with segments αF and αB of ClyA for 2B-TMD1 and 2B-TMD2, respectively (Supplementary Fig. 1E, middle panel; Supplementary Table 1). No residue aligns within the pore of ClyA (Supplementary Fig. 1E, middle panel). Whilst 3a-TMD1 aligns entirely with three identical residues and only two short gaps of four and five residues, 3a-TMD2 only matches with its N terminal side. The C terminal side of 3a-TMD2 overlaps (four residues) with αC of ClyA. 3a-TMD3 only aligns with nine residues in the αG region. Highlighting the overlaps of 2B and 3a in the structural models of ClyA indicates that none of the overlaps are with the TM sections of ClyA (Supplementary Fig. 1E, lower panel).
Summarizing the results, there is alignment of the TMDs of 3a with pore-lining domains of the ligand-gated channels nAChR and ELIC. The two proteins p7 and 2B align with pore-lining domains of the MscL, 2B-TMD1 with GluA2. The potassium channels align to some extent with all three viral proteins. No alignment is found with the membrane domains of ClyA.
Discussion
In this study, the amino acid sequences of the TMDs of viral channel proteins are aligned with the full-length sequences of host ion channel proteins and several toxins. The matching sequences of the host and toxin proteins are highlighted in the respective crystal structures. There is an emphasis on the alignment of the TMDs of the proteins since gating, ion flux, and selectivity is mostly modulated by the pore-lining TMDs.
The interpretation of the data is driven by the idea that the sequence of the viral TMD should as much as possible overlap with the target sequence together with a minimum number of gaps and respective number of amino acids in the gaps. Also, the match is favored if other residues (e.g., F,Y,W,P,S,T) than hydrophobic residues (I,L,V,A) and glycine show identical positions. The importance of the hydrophobic stretch to match is not to be underestimated. It is shown that small binding energy changes of a few kcal/mole decide between a strong binding or a weak assembly of TMDs (Yin et al., 2007). For nAChR, a hydrophobic stretch within the TMD is envisioned to play a key role in gating (Miyazawa et al., 2003).
For 3a, at this stage of the investigation the question arises: Which of the TMDs would be pore lining? The present data suggest that TMD2 could be the predominant pore-lining domain, since it aligns with the M2 domains of two chains of nAChR to a high degree. TMD3 has a very high degree of alignment only with M2 of one chain.
In a qualitative approach, and by adding previously derived information (Fischer and Hsu, 2011), the results allow the following conclusion to be drawn: Viral proteins with (i) a single TMD, as in Vpu, match with the pore-lining parts of unselective toxins like ClyA (Fischer and Hsu, 2011); (ii) two TMDs, as in p7 and 2B, overlap with mechanosensitive channel MscL, and (iii) three TMDs overlap with ligand-gated channels such as nAChR and ELIC.
The findings allow the following speculations
In a related study on Vpu—aligning its TM sequence with those of similar host proteins, as mentioned in the present study—and ClyA (Fischer and Hsu, 2011), it is shown that the TMD of Vpu matches with the pore-lining TM section of ClyA. A result from this study is that, in terms of conductance, Vpu may be seen as an unspecific conductor adopting a rather smaller pore diameter than ClyA. Together with experimental evidence (Mehnert et al., 2008), it is suggested that Vpu is not very selective and may act in dual fashion: as pore or weakly selective channel.
Proteins p7 and 2B seem to be linked to the mechanosensitive channel according to the present study. The hypothesis is put forward whether these two channels may indeed be sensitive to changes in the stress profile of a lipid bilayer. Up until now, no investigations have been launched to address this possibility. Assuming the relation to be true, one may ask at what circumstances the mechanical profile of the membrane changes. One possibility of changing the mechanical profile is during vesicle formation, which may activate the protein.
Protein 3a acts like a ligand-gated ion channel with respect to its TMDs. Again this is not unexpected considering the number of TMDs per subunit. Even though the presence of 3a is seen as a help, rather than as essential for viral survival (Narayanan et al., 2008), and interaction with host factors is not yet fully proven, current experimental evidence confirms its channel function. As a tetrameric channel, 3a channel shares the same stoichiometry as NMDA (N-methyl D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) channels (Mori and Mishina, 1995; Nakagawa, 2010; Rao and Finkbeiner, 2007). Selectivity and gating behavior of 3a still need to be evaluated. Could 3a eventually be the first viral ligand-gated ion channel (vLGIC)? In the light of TM similarity, it is anticipated that 3a uses a similar mechanism as nAChR (Hilf and Dutzler, 2009; Miyazawa et al., 2003).
Conclusions
The more TMDs per monomer, the more “hostlike” and “ligand-gated–like” the viral channel acts. With just a single TMD, the proteins may form “toxinlike channels.”
Footnotes
Acknowledgments
W.B.F. thanks the National Yang-Ming University (NYMU) and the government of Taiwan for financial support (Aim of Excellence Program). C.S. acknowledges a fellowship of the Alfried Krupp von Bohlen und Halbach Foundation and the German National Academic Foundation. This work was also supported by the National Science Council of Taiwan (NSC).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.