
Abstract
Abstract
The lion's share of bacteria in various environments cannot be cloned in the laboratory and thus cannot be sequenced using existing technologies. A major goal of single-cell genomics is to complement gene-centric metagenomic data with whole-genome assemblies of uncultivated organisms. Assembly of single-cell data is challenging because of highly non-uniform read coverage as well as elevated levels of sequencing errors and chimeric reads. We describe SPAdes, a new assembler for both single-cell and standard (multicell) assembly, and demonstrate that it improves on the recently released E+V−SC assembler (specialized for single-cell data) and on popular assemblers Velvet and SoapDeNovo (for multicell data). SPAdes generates single-cell assemblies, providing information about genomes of uncultivatable bacteria that vastly exceeds what may be obtained via traditional metagenomics studies. SPAdes is available online (http://bioinf.spbau.ru/spades). It is distributed as open source software.
1. Introduction
HMP and discovery of new antibiotics are just two examples of many projects that would be revolutionized by Single-Cell Sequencing (SCS). Recent improvements in both experimental (Ishoey et al., 2008; Navin et al., 2011; Islam et al., 2011) and computational (Chitsaz et al., 2011) aspects of SCS have opened the possibility of sequencing bacterial genomes from single cells. In particular, Chitsaz et al. (2011) demonstrated that SCS can capture a large number of genes, sufficient for inferring the organism's metabolism. In many applications (including proteomics and antibiotics discovery), having a great majority of genes captured is almost as useful as having complete genomes.
Currently, Multiple Displacement Amplification (MDA) is the dominant technology for single-cell amplification (Dean et al., 2001). However, MDA introduces extreme amplification bias (orders-of-magnitude difference in coverage between different regions) and gives rise to chimeric reads and read-pairs that complicate the ensuing assembly.1 Acknowledging the fact that existing assemblers were not designed to handle these complications, Rodrigue et al. (2009) remarked that the challenges facing SCS are increasingly computational rather than experimental. Recent articles (Marcy et al., 2007; Woyke et al., 2010; Youssef et al., 2011; Blainey et al., 2011; Grindberg et al., 2011) illustrate the challenges of fragment assembly in SCS.
Chitsaz et al. (2011) introduced the E+V-SC assembler, combining a modified EULER-SR with a modified Velvet, and achieved a significant improvement in fragment assembly for SCS data. However, we (G.T. and P.A.P.), as coauthors of Chitsaz et al. (2011), realized that one needs to change algorithmic design (rather than just modify existing tools) to fully utilize the potential of SCS.
We present the SPAdes assembler, introducing a number of new algorithmic solutions and improving on state-of-the-art assemblers for both SCS and standard (multicell) bacterial datasets. Fragment assembly is often abstracted as the problem of reconstructing a string from the set of its k-mers. This abstraction naturally leads to the de Bruijn approach to assembly, the basis of many fragment assembly algorithms. However, a more advanced abstraction of NGS data considers the problem of reconstructing a string from a set of pairs of k-mers (called k-bimers below) at a distance ≈ d in a string. Unfortunately, while there is a simple algorithm for the former abstraction, analysis of the latter abstraction has mainly amounted to post-processing heuristics on de Bruijn graphs. While many heuristics for analysis of read-pairs (called bireads2 below) have been proposed (Pevzner et al., 2001; Pevzner and Tang, 2001; Zerbino and Birney, 2008; Butler et al., 2008; Simpson et al., 2009; Chaisson et al., 2009; Li et al., 2010), proper utilization of bireads remains, arguably, the most poorly explored stage of assembly. Medvedev et al. (2011a) recently introduced Paired de Bruijn graphs (PDBGs), a new approach better suited for the latter abstraction and having important advantages over standard de Bruijn graphs. However, PDBGs were introduced as a theoretical rather than practical idea, aimed mainly at the unrealistic case of a fixed distance between reads.
When Idury and Waterman (1995) introduced de Bruijn graphs for fragment assembly, many viewed this approach as impractical due to high error rates in Sanger reads.3 Pevzner et al. (2001) removed this bottleneck by introducing an error correction procedure that made the vast majority of reads error-free. Similarly, PDBGs may appear impractical due to variation in biread (and thus k-bimer) distances characteristic of NGS. SPAdes addresses this bottleneck by introducing k-bimer adjustment, which reveals exact distances for the vast majority of the adjusted k-bimers, and by introducing paired assembly graphs inspired by PDBGs. In particular, SPAdes is able to utilize read-pairs; E+V-SC used the reads but ignored the pairing in read-pairs to avoid misassembles caused by an elevated level of chimeric read-pairs.
We assume that the reader is familiar with the concept of A-Bruijn graphs introduced in Pevzner et al. (2004). De Bruijn graphs, PDBGs, and several other graphs in this paper are special cases of A-Bruijn graphs. SPAdes is a universal A-Bruijn assembler in the sense that it uses k-mers only for building the initial de Bruijn graph and “forgets” about them afterwards; on subsequent stages it only performs graph-theoretical operations on graphs that need not be labeled by k-mers. The operations are based on graph topology, coverage, and sequence lengths, but not the sequences themselves. At the last stage, the consensus DNA sequence is restored. We designed a universal assembler to implement several variations of A-Bruijn graphs (e.g., paired and multisized de Bruijn graphs) in the same framework, and to apply it to other applications where these graphs have proven to be useful (Bandeira et al., 2007, 2008; Pham and Pevzner, 2010).
In Section 2, we give an overview of the stages of SPAdes. In Section 3, we define de Bruijn graphs, multisized de Bruijn graphs, and PDBGs. Sections 4–6 cover different stages of SPAdes. In Section 7, we benchmark SPAdes and other assemblers on single-cell and cultured E. coli datasets. In Section 8, we give additional details about assembly graph construction and data structures. In Section 9, we give a detailed example of constructing a paired assembly graph. In Section 10, we further discuss the concept of a universal assembler.
2. Assembly in SPAdes: An Outline
Below we outline the four stages of SPAdes, which deal with issues that are particularly troublesome in SCS: sequencing errors; non-uniform coverage; insert size variation; and chimeric reads and bireads:
(1) Stage 1 (assembly graph construction) is addressed by every NGS assembler and is often referred to as de Bruijn graph simplification (e.g., bulge/bubble removal in EULER/Velvet). We propose a new approach to assembly graph construction that uses the multisized de Bruijn graph, implements new bulge/tip removal algorithms, detects and removes chimeric reads, aggregates biread information into distance histograms, and allows one to backtrack the performed graph operations. (2) Stage 2 (k-bimer adjustment) derives accurate distance estimates between k-mers in the genome (edges in the assembly graph) using joint analysis of distance histograms and paths in the assembly graph. (3) Stage 3 constructs the paired assembly graph, inspired by the PDBG approach. (4) Stage 4 (contig construction) was well studied in the context of Sanger sequencing (Ewing et al., 1998). Since NGS projects typically feature high coverage, NGS assemblers generate rather accurate contigs (although the accuracy deteriorates for SCS). SPAdes constructs DNA sequences of contigs and the mapping of reads to contigs by backtracking graph simplifications (see Section 8.6).
Previous studies demonstrated that coupling various assemblers with error correction tools improves their performance (Pevzner et al., 2001; Kelley et al., 2010; Ilie et al., 2010; Gnerre et al., 2011).4 However, most error correction tools (e.g., Quake [Kelley et al., 2010]) perform poorly on single-cell data since they implicitly assume nearly uniform read coverage. Chitsaz et al. (2011) coupled Velvet-SC with the error-correction in EULER (Chaisson and Pevzner, 2008), resulting in the tool E+V-SC. In this article, SPAdes uses a modification of Hammer (Medvedev et al., 2011b) (aimed at SCS) for error correction and quality trimming prior to assembly.5
Our paired assembly graph approach differs from existing approaches to assembly and dictates new algorithmic solutions for various stages of SPAdes. Thus, we will describe several variations of de Bruijn graphs, leading to construction of the paired assembly graph (covering stages 2 and 3), before describing Stage 1. Stage 4 will be presented in Section 8.6.
3. De Bruijn Graphs: Standard, Multisized, and Paired
3.1. Terminology
Since various assembly articles use widely different terminology, below we specify a terminology that is well suited for PDBGs. All graphs considered below are directed graphs. A vertex w precedes (follows) vertex v in a graph G if there exists an edge from w to v (from v to w) in G.
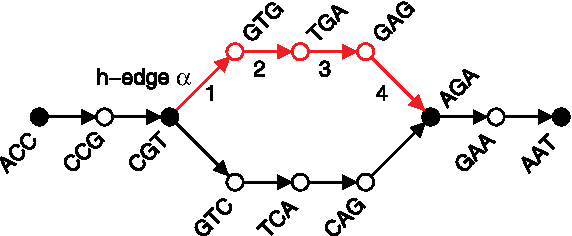
Notation for decomposing a de Bruijn graph into non-branching paths (h-paths). A de Bruijn graph on reads ACCGT
3.2. Standard de Bruijn graphs
An n-mer is a string of length n. Given an n-mer
For the rest of the paper, we fix a positive integer k. For a set R
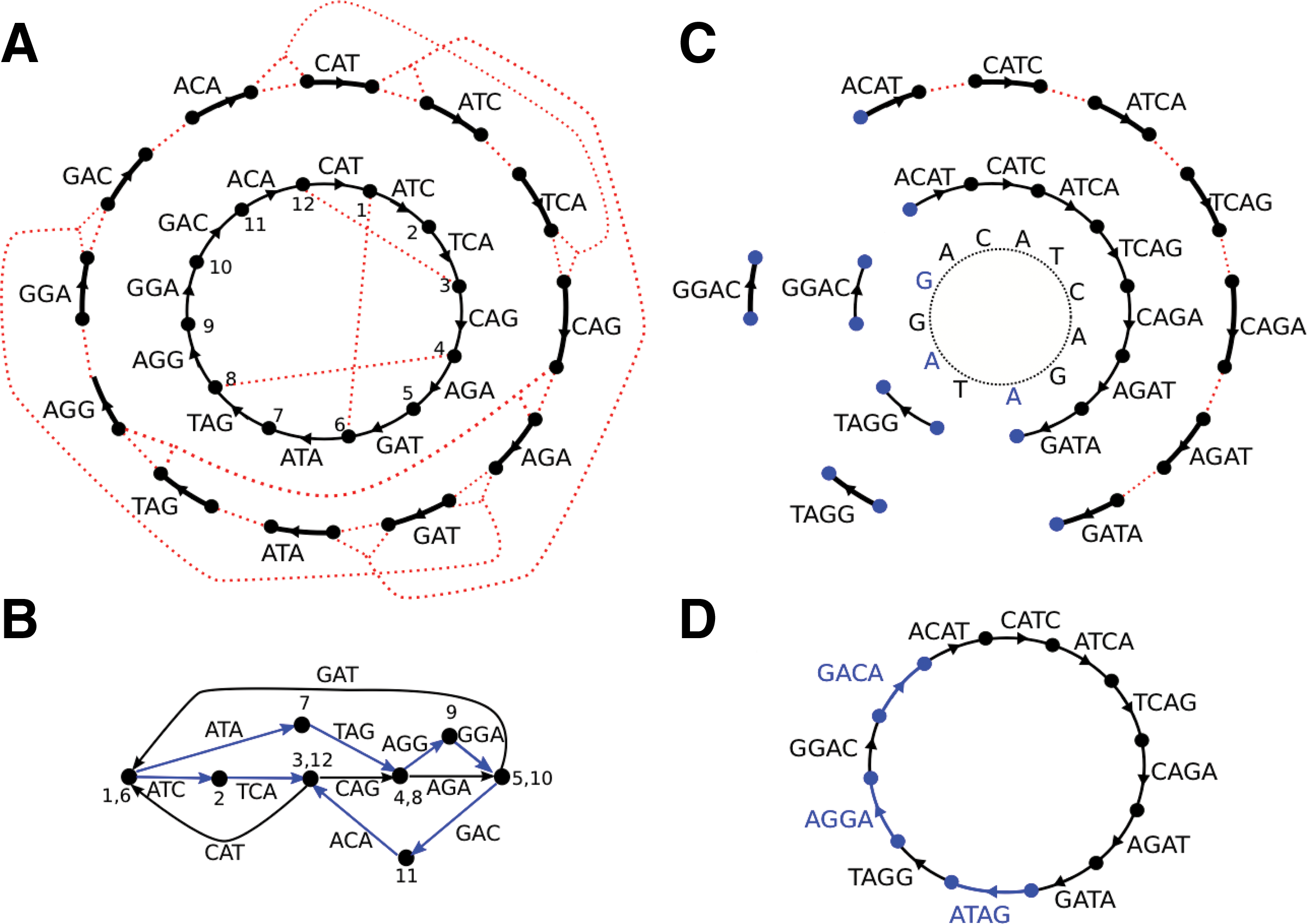
Standard and multisized de Bruijn graph. A circular G
In the de Bruijn graph DB(R
We define R
We define the coverage of an edge in the de Bruijn graph DB(R
3.3. Multisized de Bruijn graphs
The choice of k affects the construction of the de Bruijn graph. Smaller values of k collapse more repeats together, making the graph more tangled. Larger values of k may fail to detect overlaps between reads, particularly in low coverage regions, making the graph more fragmented. Since low coverage regions are typical for SCS data, the choice of k greatly affects the quality of single-cell assembly. Ideally, one should use smaller values of k in low-coverage regions (to reduce fragmentation) and larger values of k in high-coverage regions (to reduce repeat collapsing). The multisized de Bruijn graph (compare with Peng et al. [2010] and Gnerre et al. [2011]) allows us to vary k in this manner.
Given a positive integer δ < k, we define R
3.4. Practical paired de Bruijn graphs: k-bimer adjustment
In this presentation, we focus on a library with bireads having insert sizes in the range dinsert ± Δ. The genomic distance between two positions in a circular genome (and k-mers starting at these positions) is the difference of their coordinates modulo the genome length. For example, the genomic distance between a pair of reads of length ℓ oriented in the same direction with insert size dinsert is dinsert − ℓ. In the ensuing discussion, we will work with genomic distances between k-mers rather than insert sizes.
Medvedev et al. (2011a) introduced paired de Bruijn graphs (PDBG), a new approach to assembling bireads; similar approaches were recently proposed in Donmez and Brudno (2011) and Chikhi and Lavenier (2011). While PDBGs have advantages over standard de Bruijn graphs when the exact insert size is fixed, Medvedev et al. (2011a) acknowledged that PDBG-based assemblies deteriorate in practice for bireads with variable insert sizes and raised the problem of making PDBGs practical for insert size variations characteristic of current sequencing technologies.
Since it is still impractical to experimentally generate bireads with exact (or nearly exact) distances, we describe a computational approach that addresses the same goal. A k-bimer is a triple (a|b, d) consisting of k-mers a and b together with an integer d (estimated distance between particular instances of a and b in a genome). SPAdes first extracts k-bimers from bireads, resulting in k-bimers with inexact distance estimates (inherited from biread distance estimates). The k-bimer adjustment approach transforms this set of k-bimers (with rather inaccurate distance estimates) into a set of adjusted k-bimers with exact or nearly exact distance estimates. Similarly to error correction, which replaces original reads with virtual error-corrected reads (Pevzner et al., 2001), k-bimer adjustment substitutes original k-bimers by adjusted k-bimers. The adjusted k-bimer can be formed by two k-mers that were not parts of a single biread in the input data. We show that k-bimer adjustment improves accuracy of distance estimates and the resulting assembly.
4. Stage 2: Utilization of Read-Pairs in Spades
Let C be an Eulerian cycle7 in a multigraph G. The distance between edges a and b in cycle C is defined as the number of edges between S
Below we assume that reads within bireads are separated by genomic distance ≈ d0 (d0 is fixed throughout the article). The genomic distance between a pair of h-paths (i.e., strings corresponding to these h-paths) can be estimated when they are linked by bireads (that is, the first read maps to the first h-path and the second read maps to the second h-path). The estimated genomic distances between the two reads in a biread are aggregated over all bireads connecting a given pair of h-paths, through a series of transformations (Fig. 3A). The B-transformation breaks bireads into k-bimers; the H-transformation shifts the genomic distance estimate of a k-bimer connecting two h-paths to a distance estimate for the pair of h-edges representing those two h-paths, and collects this into a histogram; the A-transformation analyzes the histogram of (imprecise) distance estimates between two h-paths to determine more precise distance estimates; and the E-transformation transforms the genomic distance estimate for a given pair of h-paths into a set of k-bimers. Applying these transformations to the initial set of bireads B
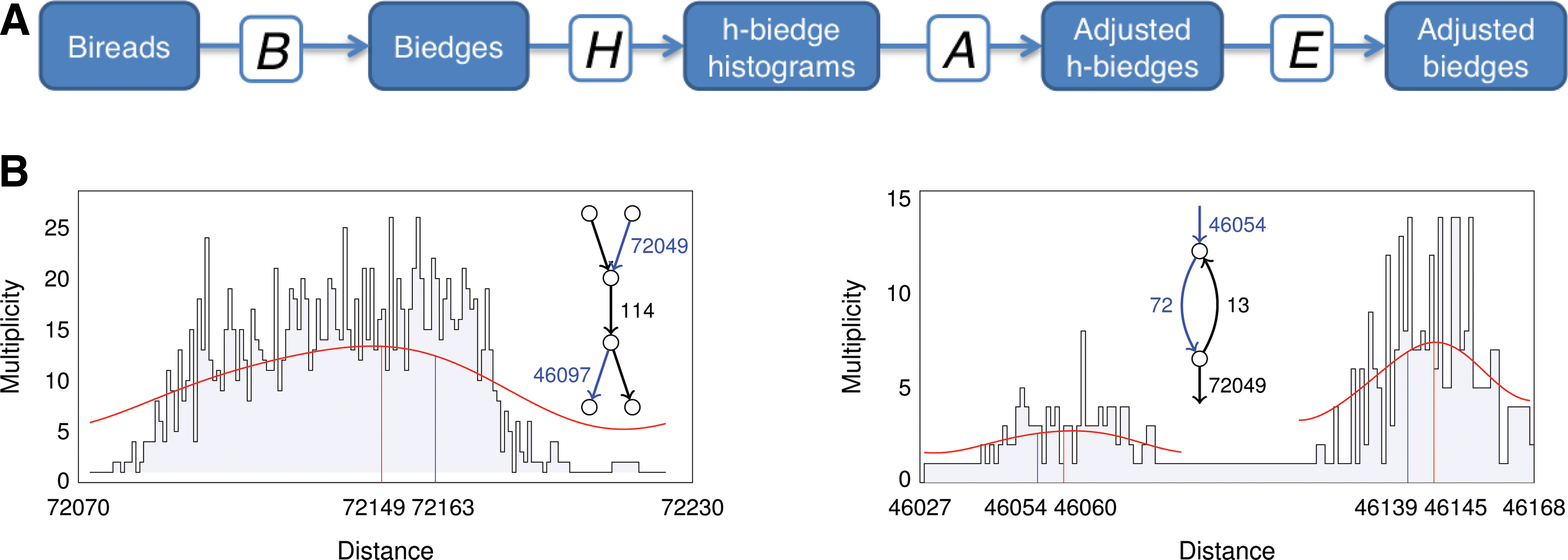
Stage 2 of SPAdes.
4.1. B-transformation
Consider a pair of reads r1 and r2 at approximate genomic distance d0 (inferred from the nominal insert length) and their mapping (described in Sec. 8.6) to paths p1 and p2 in the assembly graph. If p1 and p2 are subpaths of single h-paths in the assembly graph, we sample pairs of k-mers from these subpaths. If a k-mer a1 (resp., a2) is sampled from the read r1 (resp., r2), we form a k-bimer (a1|a2, d0 − i1 + i2), where i1 and i2 are the starting positions of a1 and a2 in reads r1 and r2. If read r1 (resp., r2) is mapped to a path overlapping with n1 (resp., n2) h-paths in the assembly graph, we sample pairs of k-mers a1 and a2 from all n1 × n2 pairs of h-paths and form k-bimers (a1|a2, d0 − i1 + i2) where i1 and i2 are the starting positions of a1 and a2 in reads r1 and r2.
Each k-bimer formed by applying the B-transformation to bireads defines a pair of edges in the de Bruijn graph, which we refer to as a biedge. Each biedge formed by edges residing on h-paths
4.2. H-transformation
Given a biedge (a|b, d), we define an h-biedge H(a|b, d) = (
4.3. A-transformation
An h-biedge histogram (α|β,*) is a multiset of h-biedges with fixed h-edges α and β and variable distance estimates. Such histograms are generated by applying H-transformations to various biedges (a|b, d) with
4.4. E-transformation
Given an h-biedge (α|β, D), we define E(α|β, D) as the set of all biedges (a|b, d) such that
Given a set B
When the distance estimate d0 between reads within bireads (and between k-mers) is exact, the transformation of bireads (and k-bimers) into h-biedges yields an equivalent representation that has no advantages. However, h-biedges have an important advantage in the case of inexact distance estimates. In this case, h-edges provide a way to adjust k-bimers and thus reveal k-mers with an exact (or nearly exact) distance estimate, making the construction of PDBGs practical. An h-biedge may aggregate distance estimates from many k-bimers into an h-biedge histogram, providing an accurate estimate for h-biedge distances. Indeed, while the number of edges (k-mers) in the de Bruijn graph is huge (millions for E. coli assembly data), the number of h-edges is rather small (a few thousand for E. coli assembly data). Huson et al. (2002), Hossain et al. (2009), and Gnerre et al. (2011) explored this aggregation to derive more accurate distance estimates in the context of scaffolding. SPAdes transforms the entire h-biedge histogram into a single adjusted h-biedge (α|β, D) (if there is a single peak in the histogram) or a small number of adjusted h-biedges (if there are multiple peaks); SPAdes further extends this approach from scaffolding (i.e., spanning gaps in coverage as in Huson et al. [2002]) to analyzing paths between h-biedges in the same connected component of the assembly graph. This analysis makes h-biedge distance estimates extremely accurate (exact for ≈ 92% of h-biedges in our single-cell E. coli dataset) and makes the PDBG framework practical.
Below, we describe further details of the A-transformation, used for analyzing h-biedge histograms.
SPAdes complements analysis of an h-biedge histogram (α|β,*) by analysis of paths between α and β in the graph to derive an accurate estimate of the distance between α and β (Fig. 3B). For example, if the analysis of the h-biedge histogram reveals a peak at distance 72149 suggesting an h-edge (α|β, 72149) but all paths between α and β have length 72163, SPAdes assigns an h-biedge (α|β, 72163) rather than (α|β, 72149). The estimation strategy must deal with cases when different paths between α and β have different lengths, when there are two or more peaks in the histogram, and when multiple paths of similar length are combined together into one peak instead of distinguishable peaks. For example, Figure 3C shows an h-biedge histogram (α|β,*) that reveals peaks at positions close to 46060 and 46146. SPAdes transforms the large multiset of individual h-biedges contributing to the histogram (α|β,*) into a single h-biedge or a small set of h-biedges. To achieve this goal, SPAdes computes the set P
5. Stage 3: From PDBGs to Paired Assembly Graphs
5.1. Biedge consistent Eulerian cycles
Let C be an (unknown) Eulerian cycle in a multigraph G. A biedge is a triple (a|b, d) where a and b are edges in G and d is an estimated distance between a and b in the cycle C. Biedges correspond to k-bimers, but this definition is sequence-independent. Given a biedge (a|b, d), we define S
A cycle C is consistent with a biedge (a|b, d) if there exist instances of edges a and b at distance d in C. Given a set BE of biedges, a cycle C is BE-consistent if it is consistent with all biedges in BE. We are interested in the following problem:
5.2. Biedge graphs
To explain the logic of constructing the paired assembly graph from the set of adjusted h-biedges A(H(B(B
For a set BE of biedges (a|b, d) with the same exact distance d, the algorithm in Medvedev et al. (2011a) for PDBGs translates into the following A-Bruijn approach to the BCEC problem:
Similarly to PDBGs, h-paths in the resulting biedge graph “spell out” paths in G shared by all BE-consistent cycles; these are typically longer than h-paths in G (as reported in Medvedev et al. [2011a], contig sizes in PDBGs significantly increase as compared to contigs in standard de Bruijn graphs).
Given a circular string G
5.3. Assembling genomes using exact h-biedges
SPAdes does not explicitly generate adjusted biedges E(A(H(B(B
We define F • If d ≥ D then • otherwise,
One can derive similar conditions for L
For a given h-biedge (α|β, D), consider all edges in the biedge graph labeled by biedges from E(α|β, D). These edges form a subpath of an h-path in the biedge graph. This subpath starts with an edge F
An illustration of this construction is shown in Figure 4, with additional details in Section 9.
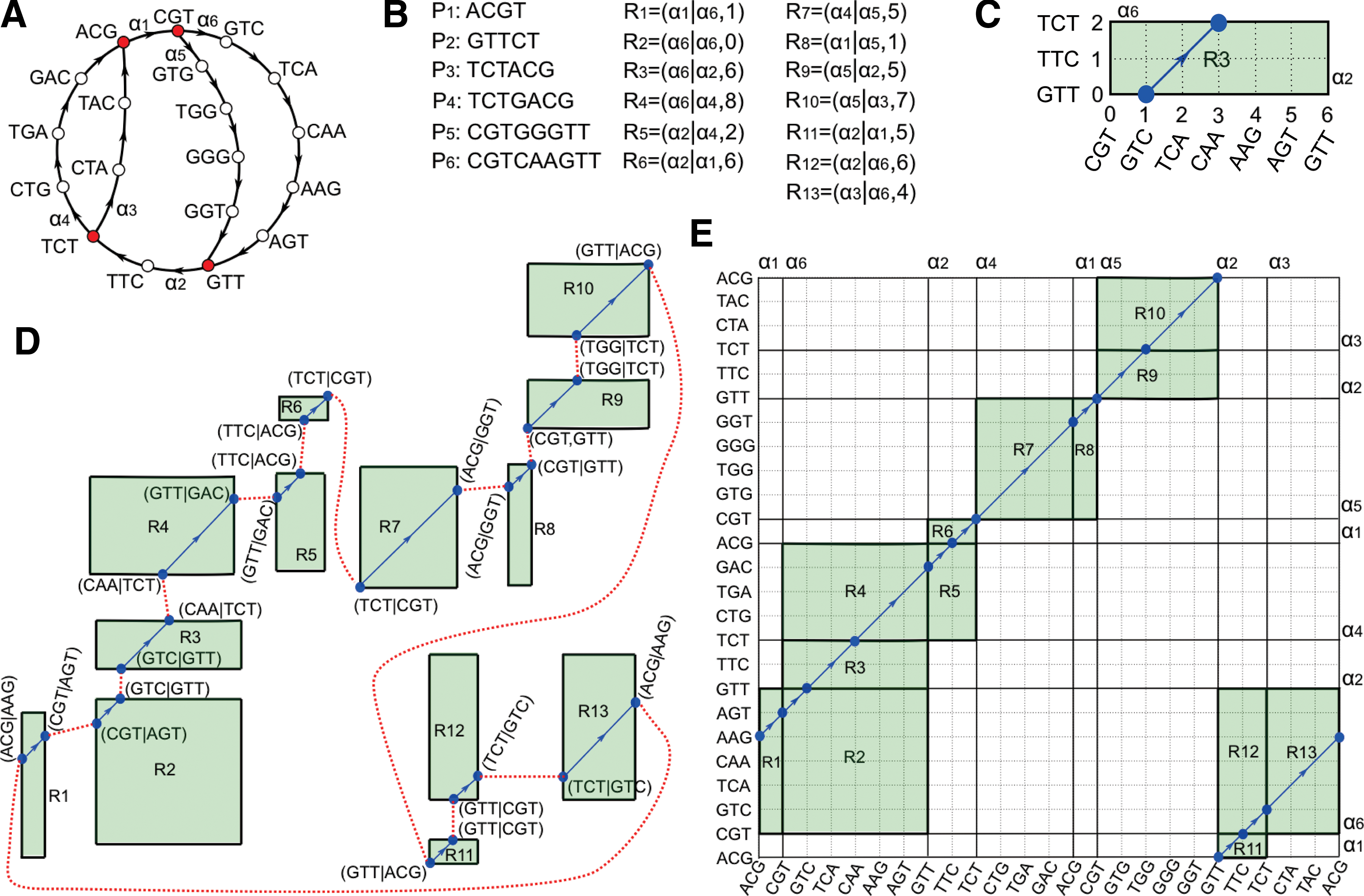
Construction of the paired assembly graph for bireads sampled from a circular 24 bp genome G
Two h-edges α and α′ in graph G are called successive if either α = α′ or α′ starts at a vertex where
While the biedge and h-biedge graphs generate long contigs for the BCEC problem with exact distance estimates, biread data generated by NGS technologies have inexact distance estimates. As Medvedev et al. (2011a) acknowledged, the advantages of PDBGs over the standard de Bruijn graphs disappear with increasing distance error Δ. Below we define a paired assembly graph addressing the case of inexact distance estimates.
5.4. Assembling genomes using inexact h-biedges
For an h-edge α, we enumerate the |
If the distance estimate D is precise, the integer points on the 45° line y = x + (d − D) define all bivertices with genomic distance d within the rectangle. The bivertex S
If for each h-biedge (α|β, D) the distance estimate D is exact, one only needs to construct the h-biedge graph as described above. However, while the vast majority of adjusted h-biedges are characterized by exact distance estimates, inexact distance estimates appear in ≈ 7% of h-biedges in our single-cell E. coli assembly and ≈ 4% of h-biedges in our multicell E. coli assembly; see Section 7.
In the case of inexact distance estimate D, the constructed 45° line shifts as compared to the correct 45° line y = x + d − D + δ (where δ is an error in D). As a result, bivertices S
We define the shift between points (x1, y1) and (x2, y2) in the rectangle as |(x1 − y1) − (x2 − y2)|. This gives a distance between the 45° lines containing these points.
Consider an h-biedge (α|β, D) where the distance estimate D has a maximal error12 δ. The incorrect distance estimate D defines an incorrect 45° line shifted by at most δ units as compared to the correct (unknown) 45° line. Therefore, the incorrect bivertices S
Conditions H1′ and H2′ below define the paired assembly graph for a set of approximate h-biedges HBE with maximal distance estimate error δ. The vertices of the paired assembly graph are multilabeled: each vertex is assigned a set of labels.
The contigs (h-paths) in the paired assembly graph are spelled out similarly to the contigs in the h-biedge graph.
6. Stage 1: Assembly Graph Construction
With respect to assembly graph construction, SPAdes develops a new idea (“bulge corremoval”) as well as modifies the gradual bulge removal approach from Chitsaz et al. (2011) and iterative de Bruijn graph approach from Peng et al. (2010).
Given a set of error corrected bireads, SPAdes starts by constructing the multisized de Bruijn graph from individual reads and computes coverage for each h-path in this graph. Using the transformation of bireads into h-biedges, SPAdes also constructs h-biedge histograms. Then SPAdes simplifies the graph by performing bulge/tip/chimeric read removals and updates the histograms to reflect all simplifications.
6.1. Multisized assembly graphs
While multisized de Bruijn graphs improve on standard de Bruijn graphs for assembly of error-free reads, they need to be modified and combined with removal of bulges/tips for assembling error-prone reads. Similarly to other assemblers, SPAdes removes bulges, chimeric reads, and tips, resulting in a simplified de Bruijn graph that we refer to as the assembly graph DB*(R
We define H-R
While one can iterate this process by taking k′ = k − 1 and increasing k by 1 at every iteration, the resulting algorithm becomes too slow. Given a set R
for
6.2. Correction of errors in assembly graphs
Errors in reads may lead to several types of structures in the de Bruijn graph:
• Miscalled bases and indels in the middle of a read typically lead to bulges (Fig. 5A). Bulges also arise from small variations between repeats in the genome. • Errors near the ends of reads may lead to tips (Fig. 5B): short, stray h-paths, with one end having total degree 1. • Chimeric reads may lead to erroneous connections in the graph, called chimeric h-paths (Fig. 5C). Chimeric h-paths may also arise from identical errors near the start of one read and near the end of another. • Input data often contains low quality reads that don't map to the genome and which may result in short, low coverage, isolated h-paths. Other assemblers may remove these based on their low coverage. We remove them after all other graph simplification procedures (since isolated h-paths are topologically distinct from bulges, tips, and chimeric h-paths, and thus may survive those steps).
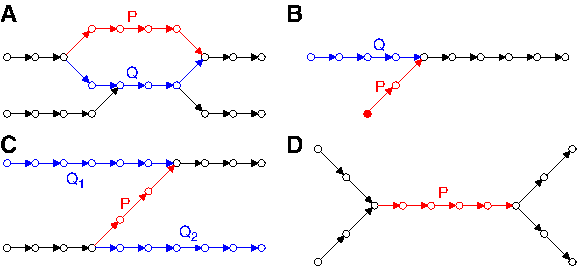
Topology of selected features within a de Bruijn graph. The red h-path, P, is the current h-path under consideration for deletion (tip removal, chimeric h-path removal) or projection to another path (bulge corremoval). The blue path(s), Q, are alternative paths. Note that other factors such as lengths and coverage are considered in addition to topology, and that the graphs continue past the regions shown.
SPAdes improves detection of these structures based on graph topology and the length and coverage of the h-paths comprising them. We also improve on how these structures are removed from the graph, and on bookkeeping that allows us to backtrack how reads map to paths in the assembly graph as a result of graph simplification.
6.3. Bulge corremoval versus bulge removal
Existing assembers often use two complementary approaches to deal with errors in reads: error correction in reads (Pevzner et al., 2001; Chaisson and Pevzner, 2008; Kelley et al., 2010; Ilie et al., 2010; Medvedev et al., 2011b) and bulge/bubble removal in de Bruijn graphs (Pevzner et al., 2004; Chaisson and Pevzner, 2008; Zerbino and Birney, 2008; Butler et al., 2008; Simpson et al., 2009; Li et al., 2010). Note the surprising contrast between these two approaches, both aimed at the same goal: the former approach corrects rather than removes erroneous k-mers in reads, while the latter approach removes rather than corrects erroneous k-mers in de Bruijn graphs. Removal (rather than correction) of bulges leads to deterioration of assemblies, since important information (particularly in the case of SCS) may be lost. SPAdes, unlike other NGS assemblers, records information about removed edges from bulges for later use before discarding them. We thus call this procedure “bulge correction and removal” (or bulge corremoval).
SPAdes uses the following algorithm for bulge corremoval. Paths P and Q connecting the same hubs form a simple bulge if (i) P is an h-path and (ii) the lengths of P and Q are small and similar. Note that Q is permitted to have intermediate vertices that are hubs, so it is not necessarily an h-path. Given a simple bulge formed by P and Q, SPAdes maps every edge a in P to an edge
SPAdes maintains a data structure (see Section 8.6) allowing one to backtrack all bulge corremovals. This is used in later stages of SPAdes to map reads to the assembly graph. For example, in Stage 2, SPAdes may correct a biedge (a|b, d) associated with an erroneous edge a by changing it into the more reliable biedge (a′|b, d) (where a was replaced by a′ through a series of projections), thus preserving (rather than removing) information about distance estimates contributed by read-pairs.
Additional conditions for removing bulges, tips, and chimeric h-paths are given in Section 8.
6.4. Gradual h-path removal
Velvet and some other assemblers use a fixed coverage cutoff threshold for h-paths in the de Bruijn graph to prune out low-coverage (and likely erroneous) h-paths. This operation dramatically reduces the complexity of the underlying de Bruijn graph and makes these algorithms practical. Since SCS projects have highly variable coverage, a fixed coverage cutoff either prevents assembly of a substantial portion of the genome or fails to cut off a significant number of erroneous h-paths. Indeed, while a short low-coverage h-path in a de Bruijn graph may appear to be a good candidate for removal if the coverage is uniform, it may represent a correct path from a low coverage region in SCS.
To deal with this problem, Chitsaz et al. (2011) proposed a specific scheduling of h-path removals that uses a progressively increasing coverage threshold. To figure out whether a low-coverage h-path is correct, E+V-SC removes some other h-paths with even lower coverage first and checks whether the h-path of interest merges into a longer h-path with an increased average coverage. This “rescuing” of lower coverage paths by higher coverage paths is a feature of SCS, since low-coverage regions are often followed by high-coverage regions.
Inspired by this approach, we implemented the even more careful gradual h-path removal strategy to improve on the algorithm from Chitsaz et al. (2011) in several respects. First, unlike E+V-SC, SPAdes iterates through the list of all h-paths in increasing order of coverage (for bulge corremoval and chimeric h-path removal) or increasing order of length (for tip removal), and updates this list as h-paths are deleted or projected (instead of deleting all edges with coverage below some threshold simultaneously as in Chitsaz et al. [2011]). This allows SPAdes to “rescue” more h-paths in the graph. Second, at some points in this process, we suspend it to run only bulge corremovals, trying to process as many simple bulges as possible. Unlike a deletion of an h-path, projection of a simple bulge is a “safer” operation because it always preserves an alternative path (of similar length) in the graph, thus allowing reads to be remapped contiguously. After a series of projections, the final contigs will correspond to the paths with highest coverage in this process. Third, SPAdes introduces an important additional restriction on h-paths that are removed that ensures that no new sources/sinks are introduced to the graph: an h-path is deleted (in chimeric h-path removal) or projected (in bulge corremoval) only if its start vertex has at least two outgoing edges and its end vertex has at least two incoming edges. (For tip removal, this only applies to the hub that is not a source or sink.) This topological condition helps to eliminate low coverage h-paths arising from sequencing errors and chimeric reads (elevated in SCS), while preserving h-paths arising from repeats (Fig. 5D). See Section 8 for additional details.
6.5. Review of all stages of SPAdes
We now briefly review all stages of SPAdes. In Stage 1, given a set of bireads, denoted B
7. Results
7.1. Assembly datasets
We used three datasets from Chitsaz et al. (2011). A single E. coli cell and a single marine cell (Deltaproteobacterum SAR324) were isolated by micromanipulation as described in Ishoey et al. (2008). Paired-end libraries were generated on an Illumina Genome Analyzer IIx from MDA-amplified single-cell DNA and from standard (multicell) genomic DNA prepared from cultured E. coli. We call these datasets SAR324, ECOLI-SC, and ECOLI-MC. ECOLI-SC and ECOLI-MC are called “E. coli lane 1” and “E. coli lane normal” in Chitsaz et al. (2011). They consist of 100 bp paired-end reads with average insert sizes 266 bp for ECOLI-SC, 215 bp for ECOLI-MC, and 240 bp for SAR324. Both E. coli datasets have 600 × coverage.
7.2. How accurate are the distance estimates for h-biedges?
The E. coli genome defines a walk in the assembly graph and a sequence of h-edges
Given a set of correct h-biedges, HBE, one can construct the h-biedge graph as described above. In practice, the set HBE is unknown and is approximated from the h-biedge histograms, resulting in the set HBE′. Below we demonstrate that HBE′ approximates the set of true h-biedges HBE well.
An h-biedge in HBE (resp., HBE′) is called a false negative (resp., false positive) if it is not present in HBE′ (resp., HBE). The false positive (negative) rate is defined as the ratio of number of false positives (negatives) to number of h-biedges in HBE′ (HBE). The false positive and false negative rates are 0.8% and 3% for the ECOLI-MC dataset and 7% and 0.7% for the ECOLI-SC dataset. Thus, SPAdes derives extremely accurate distance estimates, making the PDBG approach practical.
7.3. Benchmarking SPAdes
We benchmarked seven assemblers on three datasets (ECOLI-SC, ECOLI-MC, and SAR324). The assemblers are EULER-SR (Chaisson and Pevzner, 2008), IDBA (Peng et al., 2010), SOAPdenovo (Li et al., 2010), Velvet (Zerbino and Birney, 2008), Velvet-SC (Chitsaz et al., 2011), E+V-;SC (Chitsaz et al., 2011), and SPAdes (with and without using paired-read information). To provide unbiased benchmarking, we used the assembly evaluation tool Plantagora (http://www.plantagora.org). See Table 1.
The best assembler by each criteria is indicated in bold. EULER-SR 2.0.1, Velvet 0.7.60, Velvet-SC, and E+V-SC were run with vertex size 55. SOAPdenovo 1.0.4 was run with vertex size 27–31. IDBA was run in its default iterative mode; IDBA crashed on ECOLI-SC, so IDBA results are only shown for ECOLI-MC. SPAdes-single refers to SPAdes without Stages 2 and 3, for comparison with E+V-SC, which does not use read-pair information. SPAdes and SPAdes-single iterated over edge sizes k = 22,34,56. Statistics in this table differ slightly from statistics presented in Chitsaz et al. (2011) due to the specific criteria used in Plantagora.
Length of the largest contig without a misassembly.
The total assembly size may increase (and in some cases exceeds the genome size) due to contaminants (see Chitsaz et al. (2011)), misassembled contigs, repeats, and hubs that contribute to multiple contigs. The percentage of the E. coli genome covered filters out these issues.
Percent of genome covered is the ratio of total number of aligned bases in the assembly to the genome size.
MA: Misassemblies are locations on an assembled contig where the left flanking sequence aligns over 1 kb away from the right flanking sequence on the reference.
MM: Mismatch (substitution) error rate per 100 kbp is measured in the correctly assembled contigs.
CG: Complete genes out of 4,324 genes annotated elsewhere (http://www.ecogene.org).
Table 1 illustrates that SPAdes compares well to other assemblers on multicell and, particularly, single-cell datasets. SPAdes assembled ≈ 96.1% of the E. coli genome from the ECOLI-SC dataset, with an N50 of 49623 bp and a single misassembly. E+V-SC assembled ≈ 93.8% of the E. coli genome with an N50 of 32051 and two misassemblies. SPAdes captured ≈ 100 more E. coli genes than E+V-SC, ≈800 more than Velvet, and ≈ 900 more than SOAPdenovo.
On the ECOLI-MC dataset, the EULER-SR assembly featured the largest N50 (110,153 bp) but was compromised by 10 misassemblies. All other assemblers generated a small number of misassembled contigs, ranging from 4 (IDBA and Velvet) to 0 (Velvet-SC, E+V-SC, and SPAdes-single reads). SPAdes and Velvet also had larger N50 (86,590 and 78,602 bp) than other assemblers except for EULER-SR. All assemblers but SOAPdenovo produced nearly 100% coverage of the genome. Table 1 reveals that the substitution error rate ranges over an order of magnitude for different assemblers, with Velvet (for ECOLI-SC) and SPAdes-single reads (for ECOLI-MC) the most accurate.
We further compared E+V-SC and SPAdes on the SAR324 dataset. SPAdes assembled contigs totaling 5,129,304 bp (vs. 4,255,983 bp for E+V-SC) and an N50 of 75,366 bp (as compared to 30,293 bp for E+V-SC). Since the complete genome of Deltaproteobacterium SAR324 is unknown, we used long ORFs to estimate the number of genes longer than 600 bp, as a proxy for assembly quality (see Chitsaz et al. (2011)). There are 2603 long ORFs in the SPAdes assembly versus 2377 for E+V-SC.
7.4. Running time and memory requirements of SPAdes
Since this paper focuses on assembly (rather than error correction), below we focus on time and memory requirements of SPAdes on the ECOLI-SC reads corrected by the modified Hammer.
The running time of Stage 1 is roughly proportional to the number of iterations in the construction of multisized assembly graph. We used three iterations (k = 22, 34, 56). The time per iteration varied between 30 to 40 minutes, slightly exceeding the running time for Velvet. Stage 2 (k-bimer adjustment) took 42 minutes. Stage 3 (paired assembly graph) took 9 minutes. Stage 4 (outputting contigs) took under a minute. The total time was approximately 3 hours. Peak memory was 1.5 Gb. All benchmarking was done on a 32 CPU (Intel Xeon X7560 2.27GHz) computer.
8. Additional Details on Assembly Graph Construction
8.1. Double strandedness
To account for double-strandedness, we assemble all reads and their reverse complements together. Every edge in the graph has a reverse complementary edge, although a small number of edges may be their own reverse complement. These dual pairs of edges are kept in sync through all graph transformations.
8.2. Tip removal
Errors in the start or end of a read may lead to a chain of stray edges protruding from the assembly graph but not connecting to other reads; this is called a tip (Fig. 5B). To determine if an h-path P from hub u to hub v is a tip, we consider its topology, length, and coverage:
• Topologically, an h-path that starts with a source vertex u of outdegree 1 or ends in a sink vertex v of indegree 1 may be a tip if there is an alternative h-path Q connected to the other end. If u is a source of outdegree 1 and v has indegree at least 2, one of the other h-paths ending at v (usually there is just one other) is chosen as the alternative h-path Q. Similarly, if v is a sink of indegree 1 and u has outdegree at least 2, one of the other h-paths starting at u is chosen as the alternative h-path Q. • The length of P must be below a specified threshold, usually based on read length. For Illumina reads of length 100, we use a maximum tip length of 100. This filters out stray h-paths from bad read ends while retaining long contigs that terminate in a source or sink. • There are two coverage thresholds, one absolute, the other relative. The average coverage of P must be below a specified threshold; we used ∞ for the datasets in this paper to disable it. The ratio of the coverage of P divided by the coverage of Q must be below a threshold; we used ∞ for the ECOLI-SC dataset to disable it, and 2.5 for ECOLI-MC.
SPAdes iterates through all h-paths of the graph in order of increasing length (terminating at the maximum tip length threshold). an h-path satisfying the above criteria to be a tip (short, has an alternative h-path, and satisfies the coverage thresholds) will be deleted from the graph. When it's deleted, the hub to which it was joined may become a vertex with indegree 1 and outdegree 1, requiring us to recompute the h-paths in that portion of the graph. As with other graph simplification procedures, we update the list of h-paths on the spot. This allows us to remove all tips from the graph in one pass, while removing as few nucleotides as possible.
8.3. Gradual chimeric h-path removal
Chimeric h-paths arise from chimeric reads and from chance short overlaps between reads.
We use a variety of tests to determine if an h-path P from hub u to hub v may be chimeric. Basic gradual h-path removal considers three criteria:
• Topologically, an h-path may be chimeric if • The length of P must be below a specified threshold. For k = 56, we used maximum path length 150. • The coverage should be below a threshold, based on the library. We used 30 for ECOLI-SC and 50 for ECOLI-MC.
Since coverage varies widely within SCS datasets, some chimeric junctions may be amplified in the reads. We thus developed additional heuristics to delete some chimeric junctions with high coverage, based only on topology and length rather than coverage. (However, there may still be chimeric junctions not detected by these heuristics.) Most chimeric h-paths are observed to be smaller than ≈ k + 10. Bacterial genomes typically have a small number of long repeats (several kb long). As an example, consider a vertex v with two incoming h-paths (P0 of length ≈ k and P1 of length ≈ 3k), and one outgoing h-path (P2 of length several thousand bases); these are the only h-paths incident with v. If all three h-paths are correct, then P2 must be a long repeat of multiplicity at least 2. However, P0 satisfies the topology and length conditions for a chimeric h-path, and it is more likely that P0 is chimeric but the chimeric junction was amplified, so we delete P0.
8.4. Gradual bulge corremoval
Paths P and Q connecting the same hubs form a simple bulge if (i) P is an h-path and (ii) the lengths of P and Q are small and similar. Note that Q may have additional hubs along the path, so Q is a path but not necessarily an h-path.
By default, the length of P (and Q) is small if it is at most 150 and the lengths |P|,|Q| are similar if either
To correct a bulge, SPAdes removes path P from the graph, and substitutes each edge a of P by an edge
8.5. Isolated h-path removal
After all other graph simplification procedures, we remove isolated h-paths with length below 200.
8.6. Backtracking edges relocated during graph simplification
Although the graph simplification algorithm is universal, bookkeeping operations are performed that allow us to map reads back to their positions in the assembly graph after graph simplification. This is used in Stage 2 to map bireads to the simplified graph, and may be output in Stage 4 for downstream applications. The bookkeeping details presented here are specific for assembling a de Bruijn graph from reads and simplifying the graph. The details may change for other A-Bruijn graph applications.
For a de Bruijn graph with k-mer edges, the graph editing operations can be described as either projecting one k-mer x onto another, y, or deleting x. We keep track of this through functions M • Initially, we set M • To project edge x to edge y, where y has not yet been remapped (that is, M • To delete edge x, we set M • A series of projections during graph simplification may result in M
While the presentation in this article used edges representing k-mers, many steps in the assembler are implemented in terms of condensed edges representing sequences of varying length. Each h-path in the assembly graph (consisting of many vertices and edges) is represented as one condensed edge. As the graph is simplified, some condensed edges need to be combined into longer condensed edges.
The “universal graph” properties associated with condensed edge e include a numeric edge ID; the edge ID of the conjugate edge (for double-stranded assembly); the length (number of edges in the standard de Bruijn graph that were condensed to give this one edge; this is equivalent to length in nucleotides, ignoring the length of the terminal vertex); average coverage along e; and h-biedge histograms.
Each condensed edge e has a numeric edge ID and a string E (1) Build the de Bruijn graph and compute the set S of all k-mers. (2) Build the condensed de Bruijn graph. While forming the h-paths, we create bookkeeping structures M • Define M • Define E • Set E (3) For bulge corremoval, k-mer x is projected to k-mer y by setting M (4) Condensed edge e is deleted by setting E (5) After projection and deletion operations, vertices previously classified as hubs may change to non-hubs ( • For • Define E but overlap the final (k − 1)-mer of each string with the initial (k − 1)-mer of the next string. • For each k-mer x in E (6) After all graph simplifications, • Compute M • Compute E • If y is deleted (e = DEL) then return that x is deleted. • Otherwise, return that x is represented by k-mer y, located on edge e at offset i.
After graph simplification, we can locate where any read is represented in the graph by breaking it into its k-mers and applying the M
9. Example of Constructing Paired Assembly Graph
We illustrate the construction of the paired assembly graph in Figure 4. We treat the case of all reads at a fixed genomic distance. This small example illustrates the definitions, rather than covering all the complexities that may arise.
Read-pairs sampled from a circular 24 bp genome
form a set of single reads referred to as R
For k = 4, read ACGTCA contributes vertices ACG, CGT, GTC, TCA and edges ACGT, CGTC, GTCA to the de Bruijn graph DB(R
The de Bruijn graph DB(R
Each biread from G
The h-biedge (α6|α2, 6) results in rectangle R3 (Fig. 4C) with sides P6 and P2 and 45° line segment y = x + (d − 6) = x + (5 − 6) = x − 1 from point (1, 0) to (3, 2). Point (1, 0) in R13 is labeled by bivertex (GTC|GTT) formed by vertex 1 in path P6 and vertex 0 in path P2. Formally, it is computed as S
Applying gluing rule H2 to the 26 bivertices arising from these 13 rectangles (h-biedges) forms a single cycle that reconstructs G
In Figure 4C, all possible h-biedges are represented as rectangles on a 24 × 24 grid, with x- and y-axes corresponding to G
10. Universal Genome Assembly
In this section, we present an abstraction for assembly graphs (and other A-Bruijn graphs) for which edge labels (e.g., substrings of the genome) are hidden and only lengths and/or coverages of the edges are given. For the sake of simplicity, we address the case of a unichromosomal circular genome corresponding to a cycle in the graph. The goal is reconstruct the cycle (without using edge labels) and afterwards “translate” this cycle into a genome (by recalling edge labels).
A weighted directed graph G = (V, A, ℓ) consists of the set of vertices V, the set of (directed) edges A ⊂ V × V, and a function
Notice that a vertex/edge may appear multiple times in a path/cycle. Hence, here we distinguish vertices/edges and their instances. Depending on the context, we will treat a path/cycle as a linear/cyclic sequence of vertex/edge instances visited by the path/cycle in order.
• the subpath S • the C-distance dC(x, y) as the length of S
Let β,γ ≥ 0 be parameters specifying constraints on parallel paths and bulges in graph G.
The Universal Genome Assembly Problem (UGAP) can be formulated as follows.
Input.
• a weighted directed graph G = (V, A, ℓ); • a set BE of biedges (a|b, D), where a and b are edges in G and D is the estimated genomic distance between them; • a parameter δ ≥ 0 (maximal relative variation in distance estimates); • parameters β, γ ≥ 0 (specifying constraints on parallel paths and bulges).
Output. A shortest cycle C in G such that
• for any edge • for any biedge
Below we describe some properties of an (unknown) cycle C representing a solution to the UGAP problem that naturally guides its reconstruction; the proofs are omitted.
We define L
Example of parallel paths and bulges. Edges are labeled as vectors and vertices are labeled as scalars.
The second property ensures that the cycle C cannot be shortened by rerouting some of its subpaths:
For example, in Figure 6, if C passes through edges
The third property ensures that cycle C obeys the prescribed distances for biedges in BE:
• if • if if
if
11. Conclusion
Our results demonstrate that while SCS fragment assembly has great promise, the potential of NGS data for SCS has not yet been fully utilized. While SPAdes improves on the state-of-the-art E+V-SC assembler for SCS, it is just an initial step towards analyzing more complex SCS datasets. Several breakthroughs in single-cell genomics in 2011 have opened the possibilities of performing genome-wide haplotyping (Fan et al., 2011), studying heterogeneity within stem cell and tumor populations (Dalerba et al., 2011), tracing tumor evolution (Navin et al., 2011), and characterizing a single-cell transcriptome (Islam et al., 2011). While this article is limited to bacterial sequencing, the goal is to extend SPAdes for assembling structural variations in human SCS projects.
Footnotes
Acknowledgements
This work was supported by the Government of the Russian Federation (grant 11.G34.31.0018). S.P., G.T., and P.P. were partially supported by a grant from the National Institutes of Health (grant 3P41RR024851-02S1).
Disclosure Statement
No competing financial interests exist.
1
Chimeric reads are formed by concatenation of distant substrings of the genome, and chimeric read-pairs are formed by reads at a distance significantly different from the insert length, as well as by read pairs with an incorrect orientation.
2
This is a generic term that includes mate-pairs and paired-end reads.
3
Without error correction, de Bruijn graphs for low-coverage assembly projects with Sanger reads were too fragmented.
4
While some assemblers have built-in error correction procedures (e.g., ALLPATHS [Butler et al., ![]() ]), others do not (e.g., Velvet [Zerbino and Birney, 2008]).
]), others do not (e.g., Velvet [Zerbino and Birney, 2008]).
5
For single-cell datasets, coupling Hammer with SPAdes resulted in an improved assembly as compared to coupling EULER error correction with SPAdes. For multicell datasets, Quake and Hammer produce similar results.
6
Internally, SPAdes condenses h-paths into single edges at some stages; for presentation purposes, however, we use the uncondensed de Bruijn graph.
7
Since multiplicity of edges in graphs arising from assembly data is often unknown, assembly algorithms do not explicitly find Eulerian cycles. However, we use the Eulerian assembly framework (Idury and Waterman, 1995; Pevzner et al., ![]() ) for simplicity. An alternative formulation is to consider Chinese Postman cycles.
) for simplicity. An alternative formulation is to consider Chinese Postman cycles.
8
This condition is satisfied if
9
10
While SPAdes does not explicitly compute the E-transformation, it is useful for presenting the idea of the paired assembly graph; otherwise, the logic of the paired assembly graph construction may appear cryptic.
11
h-biedges (α|β, D) and (α|β, D′) with the same α and β but different distances correspond to different rectangles. If D and D′ are very close, the corresponding h-biedges will form parallel edges in the paired assembly graph.
12
After Stage 2 of SPAdes, the h-biedge distance estimate errors δ are greatly reduced as compared to the biread distance estimate errors Δ. For the vast majority of h-biedges, δ = 0. Moreover, the maximal error in distance estimate for each individual h-biedge can be bounded and these bounds may vary across various h-biedges. However, for the sake of simplicity, we describe the paired assembly graph for the case when δ is the same for all h-biedges.
13
IDBA incorporates each h-read (contig) for smaller k-mer sizes into a final assembly without any changes. SPAdes, in contrast, does not consider such contigs as the final truth and uses all reads at each iteration of the multisized assembly graph construction. This is important since contigs for smaller k have an elevated number of local misassemblies (usually manifested as small indels) as compared to contigs for larger k. For example, reducing vertex size from 55 to 31 (default parameter) in Velvet significantly increases the number of erroneous indels. See the ![]() section for IDBA benchmarking.
section for IDBA benchmarking.