
Abstract
Abstract
The estimation of species trees typically involves the estimation of trees and alignments on many different genes, so that the species tree can be based on many different parts of the genome. This kind of phylogenomic approach to species tree estimation has the potential to produce more accurate species tree estimates, especially when gene trees can differ from the species tree due to processes such as incomplete lineage sorting (ILS), gene duplication and loss, and horizontal gene transfer. Because ILS (also called “deep coalescence”) is a frequent problem in systematics, many methods have been developed to estimate species trees from gene trees or alignments that specifically take ILS into consideration. In this paper we consider the problem of estimating species trees from gene trees and alignments for the general case where the gene trees and alignments can be incomplete, which means that not all the genes contain sequences for all the species. We formalize optimization problems for this context and prove theoretical results for these problems. We also present the results of a simulation study evaluating existing methods for estimating species trees from incomplete gene trees. Our simulation study shows that *BEAST, a statistical method for estimating species trees from gene sequence alignments, produces by far the most accurate species trees. However, *BEAST can only be run on small datasets. The second most accurate method, MRP (a standard supertree method), can analyze very large datasets and produces very good trees, making MRP a potentially acceptable alternative to *BEAST for large datasets.
1. Introduction
With respect to mathematical foundations, one area of active research is the analysis of the sequence lengths that suffice for topological accuracy with high probability, and the development of methods that are guaranteed to recover the true tree with high probability from polynomial length sequences (Cryan et al., 1998; Erdos et al., 1999a,b; Csürős and Kao, 1999; Huson et al., 1999; Nakhleh et al., 2001a,b, 2002; Warnow et al., 2001; Mossel, 2010; Daskalakis et al., 2011; Gronau et al., 2011). These methods suggest that very large trees can be estimated with high accuracy without needing very long sequences, provided that appropriate methods can be used. The sequence length requirement of maximum likelihood estimation, however, remains an open problem (the current analysis given in Steel and Székely [1999] and Steel and Székely [2002] does not show that polynomial length sequences suffice for accuracy with high probability, but this bound is probably loose, as discussed in Mossel [2010]).
New alignment and tree estimation methods have also been developed, and substantially changed the way gene trees and alignments are estimated in practice. New multiple sequence alignment methods, such as MAFFT (Katoh et al., 2005a,b), Opal (Wheeler and Kececioglu, 2007), PRANK (Loytynoja and Goldman, 2005), and SATé (Liu et al., 2009b, 2011b), produce substantially improved alignments compared to earlier methods, and new maximum likelihood (ML) phylogeny estimation methods, including RAxML (Stamatakis, 2006), FastTree-2 (Price et al., 2010), GARLI (Zwickl, 2006), and PhyML (Guindon et al., 2010), have made very large-scale maximum likelihood analysis much more accessible. Of these methods, RAxML is probably the most popular, but as shown in Liu et al. (2011a), FastTree-2 is much faster than RAxML and may be as accurate as RAxML for very large datasets. Finally, co-estimation of alignments and trees (overviewed in Loytynoja and Goldman [2009]) is also an area of active research. Most of the methods that co-estimate alignments and trees are computationally intensive and not used much in practice, as noted in Lunter et al. (2005); however, the co-estimation methods POY (Varón et al., 2007), BAli-Phy (Redelings and Suchard, 2005), and SATé (Liu et al., 2009b, 2011b) are in use in biological dataset analyses. Of these, BAli-Phy is the only one based on a statistical model of sequence evolution that includes indels, but it is limited to perhaps 200 sequences; POY is an extension of maximum parsimony to include a gap cost and can analyze datasets with a few hundred sequences (though its performance relative to two-phase methods that first align and then estimate a tree is debated (Ogden and Rosenberg, 2007; Lehtonen, 2008; Liu et al., 2009a; Liu and Warnow, 2012); and finally, SATé can analyze datasets with thousands of sequences, and produces more accurate trees and alignments than standard approaches.
Thus, in the last 10 years or so, there has been a dramatic improvement in methods for estimating gene trees and sequence alignments. However, species tree estimation represents an additional challenge, because no individual gene tree is necessarily a good estimate of the true species tree. That is, for a number of different biological reasons, including incomplete lineage sorting (ILS) (also known as deep coalescence), gene duplication and loss, and horizontal gene transfer, gene trees can differ from the species tree (Maddison, 1997). As a result, species tree estimations need to take causes of discord between gene trees and species trees into consideration, in order to produce highly accurate estimates of the species tree.
In this article, we consider the problem of estimating species trees from estimated gene trees when the true gene trees can differ from the true species trees due to incomplete lineage sorting. Because of the frequency of deep coalescent events in phylogenetic analyses of closely related species, many methods for estimating species trees from gene trees or gene sequence alignments have been developed that explicitly take deep coalescence into account; see Degnan and Rosenberg (2009) for a relatively recent survey of methods and Edwards (2009) for a discussion of the importance of these methods for biological data analysis. Studies evaluating these methods have examined performance with respect to tree error and computational requirements on simulated datasets (Yang and Warnow, 2011), all restricted to datasets in which all gene trees have at least one individual for each species. Most of these studies have shown that methods that explicitly use statistical models to inform the estimation produce the best results; however, Yang and Warnow (2011) showed that some very simple fast methods (in particular, the greedy consensus) came close to the accuracy of a statistically based method, BUCKy (Larget et al., 2010) on tree distributions estimated using MrBayes (Ronquist and Huelsenbeck, 2003).
We focus our attention on the problem of estimating species trees from incomplete estimated gene trees, by which we mean the case where the gene trees might not contain any individuals for some species. In this case, methods that require that all the gene trees have the same set of taxa (such as the greedy consensus and BUCKy) cannot be applied. In addition, results from prior studies that evaluated methods on inputs in which all gene trees have at least one individual from each species are not necessarily applicable, since performance on incomplete gene trees could be different.
We begin with a study of the Minimize Deep Coalescence (MDC) problem introduced in Maddison (1997). This problem takes as input a set of rooted binary gene trees, each on the same set of taxa, and seeks the species tree for which there is a minimum total number of deep coalescences. Although this approach to species tree estimation is not statistically consistent when gene trees can differ from the species tree due to ILS (Than and Rosenberg, 2011), it is one of the most popular techniques for estimating species trees when ILS is suspected. We show how to extend MDC to the case where the gene trees are incomplete, and we prove that Phylonet-MDC (Than et al., 2008) solves this computational problem exactly. We then report on a simulation study we performed to evaluate methods for estimating species trees from incomplete gene trees or alignments for datasets with multiple genes and with 11, 17, or 100 taxa. We compare *BEAST (Heled and Drummond, 2010), a Bayesian method for estimating species trees from gene sequence alignments when genes can differ from species trees due to ILS, to methods based on MDC (iGTP-MDC [Chaudhary et al., 2010] and Phylonet-MDC). We also make comparisons to a heuristic for MRP (matrix representation with parsimony, a standard supertree method) (Baum, 1992; Ragan, 1992) known to be one of the most accurate supertree methods (Swenson et al., 2010; Kupczok et al., 2010; Swenson et al., 2011b) and to heuristics to minimize duplications or duplications + losses in iGTP (Chaudhary et al., 2010), none of which consider ILS when estimating species trees. We compare these methods on datasets simulated on gene trees that can differ from species trees due to ILS and report the missing branch rates of each species tree that we compute.
Although we did not attempt to run *BEAST on the 100-taxon datasets (due to its excessive computational requirements on large datasets), it produced the most accurate trees on the datasets with 11 or 17 taxa. Comparisons between other methods showed that generally MRP gave the most accurate results, and that (when it could be run), the exact version of Phylonet-MDC produced the next most accurate results. In addition, MRP was very fast on these datasets, producing results in under a minute on all datasets. These results suggest that at least for some conditions involving incomplete gene trees, methods that attempt to solve MRP may be computationally tractable ways of producing reasonably accurate species trees, and perhaps better than methods that optimize the MDC criterion. However, for those datasets for which statistical methods (such as *BEAST) can be run, they may be able to produce substantially more accurate trees than all other methods. (See Table 1 for a list of acronyms used in this article and their definitions, and Table 2 for a list of software used in the simulation study.)
2. Theoretical Results for MDC
We begin by defining the MDC problem in the context of complete rooted, binary gene trees. We then show how to extend MDC to incomplete gene trees.
2.1. MDC for complete gene trees
The MDC problem is as follows:
• Input: A set • Output: a rooted, binary species tree T that minimizes the number of extra lineages with respect to
To define the MDC problem, therefore, we need to define XL(T, ti), i.e., the number of extra lineages of a species tree T with respect to a gene tree ti. Visually, this is defined by embedding the gene tree ti into the species tree T, and then counting how many lineages there on each edge of the species tree; for a given edge, the number of extra lineages is one less than the total number of lineages on the edge (Maddison, 1997). This visual definition of the MDC cost is not necessarily easy to understand.
An alternative definition is given in terms of what are called “B-maximal clusters,” which we now define. A cluster is a subset of the leaf set of a rooted tree, consisting of all the leaves below some internal node. Thus, given a cluster within a tree defined by the node v, we can also define its parent cluster to be the cluster associated with the parent of v. Furthermore, the “parent edge” of a cluster C defined by the node v (i.e., v is the root of the subtree whose leafset is C ) is the edge e = (v, w), where w is between v and the root of t. Let T and t be rooted binary trees on S, with T denoting the species tree and t denoting the gene tree. Let B be a cluster of T . We will say that cluster A of t is B-maximal if A ⊆ B and the parent cluster for A is not a subset of B.
For a cluster B of T , we define kB(t) to be the number of B-maximal clusters of t, and we let wB(t) = kB(t) − 1. It is now known that the embedding of the gene tree t into the species tree T that maps every node in t to MRCA (most recent common ancestor) in T of the leafset below v optimizes the MDC cost. Furthermore, for this embedding, the number of lineages “leaving” the parent edge of the cluster B (i.e., the edge between B and the root of the tree T) is kB(t); therefore, the number of extra lineages on the parent edge is wB(t) (one less than the number of lineages) (Yu et al., 2011). Note that wB(t) ≥ 0 since t and T have the same set of taxa, and that XL(T, t) = ∑ B w B (t), where the sum is taken over all clusters B in the tree T, is the number of extra lineages implied by the pair t,T. This is what is meant by the MDC cost for T with respect to gene tree t.
The MDC problem can then be restated as follows:
• Input: set • Output: binary rooted species tree T on S such that
Than and Nakhleh (2009) noted that this problem could be solved exactly by finding a minimum weight clique of size n − 2 in a graph in which there is a node for every possible cluster in the species tree (i.e., subset of taxa), an edge between nodes where their clusters are compatible (meaning that they can co-exist in a rooted tree), and where the weight of the node for cluster B is ∑ i w B (ti). This observation yielded the exact version of Phylonet-MDC (Than et al., 2008). By restricting the set of nodes to those clades that appear in the input set of gene trees, they produced the heuristic version of Phylonet-MDC; this method solves the MDC problem exactly when constrained to species trees whose clades are drawn only from the input gene tree clades. Finally, Yu et al. (2011) showed how to modify the Phylonet-MDC algorithm so that it could work with unrooted, partially resolved gene trees and find optimal rooted refinements and species trees that minimize the MDC score.
2.2. Extension to incomplete gene trees
We now discuss how to extend the MDC criterion to handle incomplete gene trees, where the gene tree leaf sets may not contain all the species. We begin with a definition: If S is the full set of taxa and t is a binary rooted tree on a subset of S, then we say that t′ is a completion of t if t′ is a binary rooted tree that contains all the taxa in S and that agrees with t when restricted to the taxa in t. Thus, a completion t′ is obtained by adding additional leaves to t so that it contains all the taxa it is missing. With this, we can now define MDC for incomplete gene trees.
• Input: set • Output: binary rooted species tree T and completions
We will refer to this problem as MDC-incomplete, and we will denote a solution to MDC-incomplete on input set
Recall that kB(t) is defined for the case where the gene tree t is rooted and has the same set of taxa as the species tree; in this case, it equals the number of B-maximal clusters of t. Furthermore, again for the case where the gene trees all have the same set of taxa, we have defined • wB(t) = 0 if • wB(t) = kB(t) − 1, otherwise.
In other words, we generally use the same definition for wB(t), except when B is entirely disjoint from the leafset of t. This definition ensures that wB(t) ≥ 0 for all clusters B and all gene trees t.
• Input: set • Output: binary rooted species tree T on S = ∪
i
S
i
such that ∑
i
∑
B
w
B
(ti) is minimized, where i ranges from 1 to k and B ranges over all clusters in T.
We refer to this problem as MBMC-incomplete, and the optimal tree given input
The main result in this article is the following:
Theorem 1
Let
In other words, the species tree T that optimizes the MBMCinc criterion is the species tree component of the optimal solution to MDCinc.
Phylonet-MDC and iGTP-MDC
The software packages Phylonet (Than et al., 2008) and iGTP (Chaudhary et al., 2010) handle incomplete gene trees differently when attempting to solve MDC, in that they can compute MDC scores differently. In particular, Phylonet defines the MDC score using the MBMCinc cost, as described above (i.e., the cost of a species tree T is ∑
i
∑
B
w
B
(ti), where B ranges over the clusters in T and i ranges from 1 to k). Theorem 1 thus shows that Phylonet-MDC computes the MDC score correctly. By contrast, there are inputs for which iGTP-MDC does not return this score, indicating that iGTP-MDC defines the MDC score differently for incomplete gene trees. One particular instance in which this occurs is as follows:
• Input gene trees: T1 = (((a, b), c), d), T2 = ((b, c), (d, e)), and T3 = ((a, d), (b, e)). • Output of Phylonet-MDC exact version: ((d,e),(a,(b,c,))), claiming 3 extra lineages. • Output of iGTP(MDC): ((d,e),(c,(a,b))), and claims 2 extra lineages.
By our calculation and definition for MDCinc, Phylonet-MDC correctly computes the number of extra lineages, but iGTP does not. We conjecture that iGTP seeks the species tree T that minimizes ∑ i XL(Ti, ti), where Ti is the subtree of T induced by Si. Therefore, iGTP-MDC and Phylonet solve different problems when given gene trees that are incomplete.
3. Establishing the Relationship Between MDCinc and MBMCinc
In this section, we establish the relationship between optimizing the MDCinc and MBMCinc problems. As a result of this theorem, it will follow that the exact formulation of Phylonet-MDC solves the MDC problem optimally. That is, given an input of incomplete, binary rooted gene trees, to find an optimal species tree and completions of the binary gene trees it will suffice to find a minimum weight clique containing n−2 vertices (where n is the number of taxa) in the graph defined by Phylonet, which has one vertex for each possible cluster, edges between vertices exist if and only if their clusters are compatible (either disjoint or one contains the other), and the weight on the vertex for cluster B set to wB.
We begin with the following lemma.
Lemma 1
Let T and t be rooted binary trees with
Proof
We consider the four cases that can occur in a species tree T in which B, B0 and X are clusters:
• Case 1: B ⊆ X • Case 2: B ⊆ B0 • Case 3: B0 ∪ X ⊆ B • Case 4: (B0 ∪ X) ∩ B = ∅
We take each case in turn.
Case 1: B ⊆ X. In this case, B is a cluster in the clade on X, and hence a cluster in t′. Therefore, wB(t′) = 0. Since B ⊆ X, it follows that
Case 2: B ⊆ B0. First, if
Conversely, suppose A is a B-maximal cluster in t′. Since A ⊆ B ⊆ B0, A is a cluster in t. If A is B-maximal in t, then we are done. Else, suppose A is not B-maximal in t. Note that A cannot have the same parent cluster in t and t′, since otherwise A is also B-maximal in t′ (contradicting our hypothesis), and so A's parent cluster in t′ must contain A0 ∪ X. Hence, A's parent cluster in t must be defined by an internal node on the path from the root of A0 to the root of t. Label the nodes on that path
Case 3: B0 ∪ X ⊆ B. Our first observation is that A0 is B-maximal in t if and only if A0 ∪ X is B-maximal in t′. Hence, we need only concern ourselves with the B-maximal clusters in t other than A0, and (equally) with the B-maximal clusters in t′ other than A0 ∪ X. However, when A ≠ A0, it is easy to see that A is a B-maximal cluster in t if and only if A is a B-maximal cluster in t′. Hence, wB(t) = wB(t′).
Case 4: (B0 ∪ X) ∩ B = ∅. It is easy to see that for any cluster A, A is B-maximal in t if and only if A is B-maximal in t′, and so wB(t) = wB(t′).
The following lemma is obvious and the proof is omitted:
Lemma 2
Let t be an incomplete gene tree, T a species tree, and t′ a completion of t to the taxon set of T. Then wB(t) ≤ wB(t′) for all clusters B of T.
Theorem 1
Let
Proof
Let
For the converse, let
4. Methods
4.1. Overview
The simulation study used gene sequences that evolve down gene trees that can differ from the true species tree due to ILS. To produce these sequence datasets, we used sequences used in previous studies and provided to us by the authors of these studies–the 11-taxon datasets from Chung and Ané (2011), the 17-taxon datasets from Than and Nakhleh (2009), and the 100-taxon datasets from Yang and Warnow (2011). We summarize the simulation protocols used in these studies here, and direct the reader to the relevant publication for the details of how the data were generated.
In each case, a model species tree was generated (typically using a birth-death process). Then a set of gene trees within each species tree was produced under a coalescent process, so that for each gene one individual was sampled for each species. This produces gene trees with branch lengths that can differ topologically from their associated species tree due to ILS. DNA sequences were then simulated down each gene tree. For the 11-taxon and 17-taxon datasets, these simulations were done under a substitution-only model, and for the 100-taxon datasets these simulations were done under GTR+Gamma+gap models with varying gap lengths; thus, the 100-taxon datasets evolved with indels as well as with substitutions. Many replicates were generated for each model condition, and each replicate consisted of true sequence alignments for each gene.
For each replicate dataset, we had the true alignment as well as the true tree. We then deleted taxa randomly, varying the number of taxa removed, from each gene sequence alignment, thus producing incomplete gene sequence alignments. On each resultant gene sequence alignment we estimated trees using FastTree-2 (Price et al., 2010); this produces a tree as well as branch support estimations. We produced a 75%-branch support version of each estimated gene tree by contracting all edges with support below 75%.
For each replicate of each model, we thus have three types of datasets (each consisting of a collection of gene sequence alignments and trees): the true gene sequence alignment, the binary trees estimated by FastTree-2 on the true gene sequence alignment, and the 75%-branch support FastTree-2 trees estimated on each true gene sequence alignment.
For each such dataset, we estimated species trees using the following techniques:
• iGTP v. 1.1. We explore all three optimization criteria (deep coalescence, duplications, duplication-loss) available in iGTP. We ran iGTP on 75% support version of the input binary trees, although it is not guaranteed to give meaningful outputs for non-binary gene trees. • Phylonet v. 2.4. We explore both heuristic and exact version of Phylonet used to solve the MDC problem on both binary and unresolved gene trees. However, the exact version can only be run on small datasets, and so we used it only on the 11-taxon datasets. • Matrix Representation with Parsimony (MRP). We ran MRP heuristics on the FastTree-2 trees (both binary and 75%-support versions), using a Python script to run a parsimony ratchet analysis using PAUP*, with 100 iterations, followed by taking the greedy consensus of the set of trees. • *BEAST v. 1.6.2. We ran *BEAST on the true alignments for each dataset using its default settings.
We recorded the average (over all replicates) missing branch rate and running time for each method. When computing the missing branch rate, we compare the estimated species tree to the subtree of the true species tree induced by those species present in at least one gene tree.
4.2. Datasets
We ran our experiments on datasets that evolve with ILS. We used 11-taxon datasets, each with 10 genes, obtained from Chung and Ané (2011). We also used 17-taxon datasets with 8 genes each, used previously in Than and Nakhleh (2009). Finally, we used 100-taxon datasets with 25 genes each, used in Yang and Warnow (2011).
5. Results
5.1. Missing branch rates
We begin by discussing performance with respect to missing branch rates.
5.1.1. Results on 11-taxon datasets
For these datasets, we were able to run the exact version of Phylonet-MDC, and hence solve the MDC problem exactly. As before, we ran the heuristic version of Phylonet-MDC, the three iGTP methods (for the MDC score, duplication score, and duplication plus losses score), and MRP. We explored results with two, three, and five missing taxa (Fig. 1)
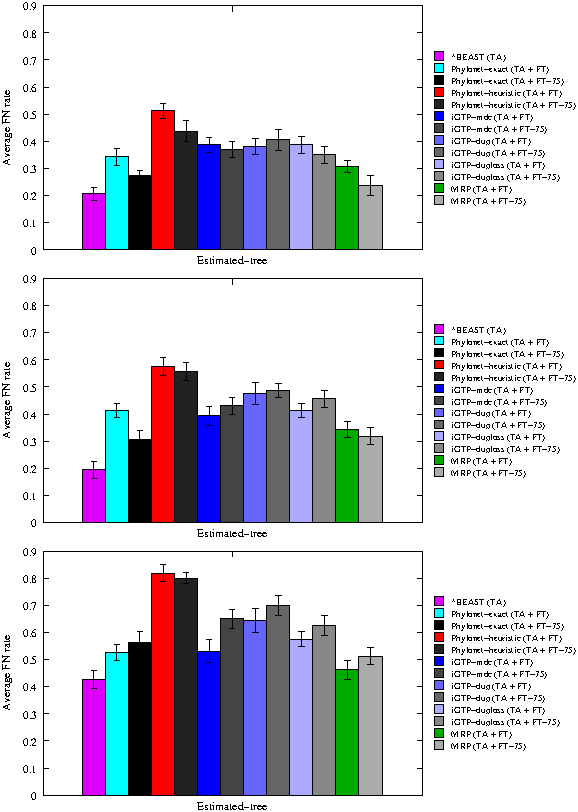
Average missing branch rates of methods on 20 11-taxon 10-gene datasets on true alignments (TA). Gene trees are estimated using FastTree-2 (FT), and in some cases the branches with support less than 75% are contracted (FT-75). From top to bottom, the number of missing taxa is 2, 3, and 5.
The first observation is that *BEAST produced the most accurate species trees, for all percents of missing taxa. The second best method varied depending on the percentage of missing taxa, with MRP on the 75%-support trees best for 20% missing taxa, Phylonet-exact on the 75%-support trees best for 30% missing taxa, and MRP best for 50% missing taxa. Thus, there was no clear second best method. Furthermore, although these three methods generally gave reasonably good results, they were not always among the next most accurate. Between the iGTP methods, iGTP-dup had the worst results, and iGTP-MDC and iGTP-duploss were sometimes reasonably accurate. A noteworthy trend was that Phylonet-heuristic gave the worst results at all percents of missing taxa, whether applied to the fully resolved trees or the 75%-support trees. Finally, using the 75%-support trees instead of the fully resolved trees improved MRP and Phylonet (both exact and heuristic) for small numbers of missing taxa, but not when the number of missing taxa was large. Also, using the 75%-support trees did not help the other methods.
5.1.2. Results on 17-taxon datasets
Performance on 17-taxon datasets with 8 genes showed similar results (Fig. 2). Because of the number of taxa, we did not run Phylonet-exact. However, the results we saw here are similar to what we saw on the 11-taxon datasets. As before, *BEAST was the most accurate, for all percents of missing taxa. The next best methods were MRP and iGTP-MDC (on either binary or 75%-support trees), and sometimes also iGTP-duploss on binary trees, but all had at least 7% higher missing branch rates than *BEAST. The worst results were obtained using Phylonet-heuristic and iGTP-dup on either the binary or 75%-support trees.
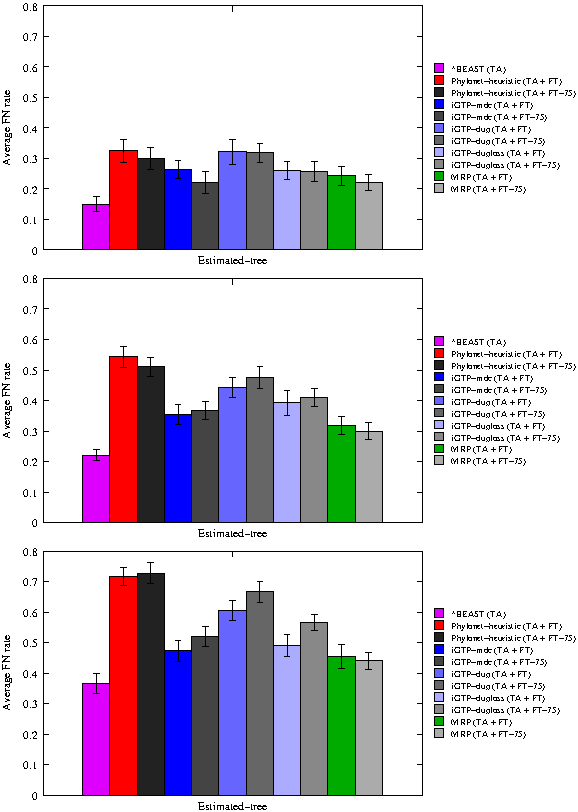
Average missing branch rates of methods on 20 17-taxon 8-gene datasets on true alignments (TA). Gene trees are estimated using FastTree-2 (FT), and in some cases the branches with support less than 75% are contracted (FT-75). From top to bottom, the number of missing taxa is 1, 5, and 8.
5.1.3. Results on 100-taxon datasets
We now describe results on the 100-taxon datasets. Because of the number of taxa, we did not run *BEAST (running long enough to reach convergence was infeasible for this experimental study), nor Phylonet-exact. However, these data allow us to compare the other methods, Phylonet-heuristic, the three variants of iGTP, and MRP, on both binary and 75%-support trees (Fig. 3).

Average missing branch rates of methods on 10 100-taxon 25-gene datasets on true alignments (TA). Gene trees are estimated using FastTree-2 (FT), and in some cases the branches with support less than 75% are contracted (FT-75). From top to bottom, the number of missing taxa is 10, 30, and 50.
On the estimated gene trees, MRP on the 75%-support trees gives the most accurate trees, but MRP on binary trees comes quite close. The least accurate method is Phylonet-heuristic on binary trees, and Phylonet-heuristic on 75%-support trees is only slightly better (and much less accurate than all the other methods). A comparison between the iGTP methods no longer shows no reliable differences: for example, sometimes iGTP-MDC is the best and sometimes it is the worst of the three.
5.1.4. Overall results
For all levels of missing data, certain trends were clearly seen. Results for all methods improved when given more estimated gene trees rather than fewer; these trends are to be expected, and consistent with prior studies (Yang and Warnow, 2011). In addition, we saw that for each species tree estimation method, the missing branch rate increased with increased levels of taxon deletion, but the increase in error was particularly large for the heuristic version of Phylonet-MDC.
The relative performance between methods showed clearly that when analyzing estimated gene trees, *BEAST produced the most accurate results (as indicated by the lowest missing branch rate). MRP, especially on the 75%-support trees, typically came in second or close to second, and when Phylonet-exact could be run, it gave results that were close to that of MRP. However, in all the experiments, Phylonet-heuristic gave the least accurate results or tied for last. Comparisons between the iGTP methods depended on the model condition, and no overall trends could be observed.
Using 75%-support trees had a variable effect on the different methods we explored. First, on low taxon deletion levels, Phylonet-MDC (in either the exact or heuristic version) and MRP were improved by using the 75%-support trees, but this changed for the highest level of taxon deletion. Why this is happened is unclear, although it could be that the branches on the estimated gene trees had low support when estimated on very sparse taxon sets (such as would be obtained by deleting many taxa), leading to more loss of information when using the 75%-support trees. On the other hand, the iGTP methods did not show any advantage when used with 75%-support trees, and were often hurt.
5.2. Computational issues
We also evaluated the running time and memory usage of the different methods we studied. Phylonet-exact uses time that is exponential in the number of taxa, and so could only be run on the 11-taxon datasets; however, on these datasets it completed in less than 2 seconds. The next most expensive method is *BEAST, which must be run long enough to converge to the stationary distribution. Therefore, we only ran *BEAST on the 11-taxon and 17-taxon datasets. On average, *BEAST finished its analyses in 15 minutes on the 11-taxon datasets and 20 minutes on the 17-taxon datasets. The remaining methods were much faster: all finished in under a second on the 11-taxon and 17-taxon datasets, and in under a minute on the 100-taxon datasets. Some differences in running time were evident on the 100-taxon datasets, where Phylonet-heuristic finished in 6 seconds, MRP finished in 20 seconds, but the three iGTP methods took between 20–64 seconds. Peak memory usage by these methods all differed, but only *BEAST used any substantial memory—about 1GB on the 17-taxon datasets.
6. Discussion
We begin with some observations about methods that attempt to optimize the MDC criterion. First, it is clear that iGTP-MDC generally gives more accurate trees than Phylonet-MDC run in its heuristic mode; however, when the exact version of Phylonet-MDC can be run, it produces more accurate trees than its heuristic version, and also more accurate trees than iGTP-MDC. The reason for this is likely due to the improved MDC scores produced by using the exact version of Phylonet-MDC (which are mathematically guaranteed), compared to the other methods. It is worth noting that the substantial reduction in topological accuracy (and MDC scores, results not shown) by using the heuristic version instead of the exact version of Phylonet-MDC is almost certainly a result of the fact that all the gene trees are incomplete, with randomly deleted taxa. This greatly impairs the ability of Phylonet-MDC's heuristic to score trees that are topologically similar to the true tree, since all the clades in any estimated tree must be drawn from the input gene tree clades in this case. However, the heuristic used in iGTP-MDC explicitly searches through treespace and so is not impaired in the same way. Given that previous research (Yang and Warnow, 2011) has shown very good trees resulting from Phylonet-MDC's heuristic version when the input gene trees are all complete, it seems likely that Phylonet-MDC might give better results when the taxon deletion is not random, or when at least some of the gene trees are based on complete taxon sets. Thus, although this study showed poor accuracy for Phylonet-MDC's heuristic, this trend may not hold under other circumstances, including those that might better represent systematic practice. Future work will investigate this possibility.
We also note that contracting low support branches in estimated gene trees typically (but not always) benefited Phylonet-MDC and MRP, but not the iGTP methods. This difference is likely due to differences in the treatment of unresolved gene trees within the iGTP, Phylonet-MDC, and MRP software. For example, it seems likely that iGTP-MDC and Phylonet-MDC do not score proposed species trees identically when the input gene trees are unresolved (Phylonet-MDC scores species trees with respect to optimal refinements of unresolved gene trees (Yu et al., 2011), a guarantee that may not be true of iGTP-MDC).
This study establishes that there is currently no computationally feasible solution for estimating highly accurate species trees from incomplete gene trees for large numbers of taxa. That is, only *BEAST was able to produce highly accurate species trees; all other methods had much higher error rates. Therefore, for small enough numbers of taxa so that *BEAST can be run properly without huge running times, very accurate species trees can be computed. Although this study did not investigate the feasibility of running *BEAST on larger datasets, other studies with Bayesian methods have shown that proper analyses of datasets (even small ones) can take weeks of analysis to reach convergence (Yang and Warnow, 2011). Therefore, the poor results of the other methods on larger datasets suggests that highly accurate species tree estimations from incomplete gene trees and alignments may be beyond what current methods can achieve.
This study also suggests some limitations to analyses based on MDC. Unsurprisingly, we saw that optimizing MDC generally gave better results than optimizing duplications or duplications and losses. We also observed (Yang and Warnow, 2011) that optimizing the total number of duplications and losses produced more accurate trees than optimizing duplications alone.
Finally, and perhaps most interestingly, we noted that optimizing MDC produced generally less accurate trees than optimizing MRP. This is a very interesting result, given that MRP is agnostic about the cause of incongruence between gene trees, and MDC explicitly addresses ILS as the cause for incongruence. However, there is no mathematical explanation for why MRP would perform well, and so this remains only an empirical observation.
This study shows that the standard heuristics (the parsimony ratchet as implemented in PAUP*) for the supertree method MRP produces highly accurate species tree estimations, even though it does not consider ILS, and can do so reasonably quickly, even on large datasets. These observations, combined with the observation that none of the methods we studied (other than *BEAST) that explicitly take into account events such as ILS or duplication and loss produced trees as accurate as MRP, suggest that optimizing MRP may be a reasonable approach to species tree estimation for large datasets, when statistical methods (such as *BEAST) cannot be run for computational reasons. Therefore, other supertree methods, such as SuperFine (Swenson et al., 2011a; Neves et al., 2012; Nguyen et al., 2012), a new supertree method that has been shown to produce better MRP scores and more accurate trees than standard MRP heuristics (while also being faster than standard MRP heuristics), should also be investigated. Finally, for complete gene trees, the greedy consensus produced highly accurate species trees, despite not being statistically consistent (at least when used on rooted gene trees, as shown in Degnan et al. [2009]). Given this observation, other consensus methods (Bryant, 1991; Kannan et al., 1995; Phillips and Warnow, 1996) might also be useful for estimating species trees for large numbers of taxa.
7. Conclusion
Species tree estimation from incomplete gene trees that can differ from the true species tree due to ILS presents many interesting theoretical and empirical challenges: excellent results can be obtained using *BEAST, a statistical approach that explicitly models the processes that cause incongruence between gene trees and species trees, but *BEAST is too computationally intensive for even moderately large datasets. In contrast, a very simple supertree method, MRP, is able to provide reasonably good results on very large datasets, even though it does not provide statistical guarantees. Thus, while it seems likely that methods based on sound statistical models will produce the most accurate species trees, the current methods that can analyze incomplete gene trees are either limited to small datasets, or are not based upon statistical models of gene tree incongruence. Given the increased use of multi-marker analyses for species tree estimation, methods that are both statistically-based and can run on large datasets (and can analyze incomplete gene datasets), are likely to have high impact. Future work will hopefully produce methods that are scalable and statistically based, and that produce highly accurate trees on datasets with large, incomplete gene trees.
Authors' Contributions
T.W. designed the study; S.B. performed the study; S.B. and T.W. proved the theoretical results, analyzed the data, and wrote the article.
Footnotes
Acknowledgments
We thank the anonymous reviewer for helpful suggestions. This research was supported by the U.S. National Science Foundation (DEB 0733029 and DBI-1062335), the John P. Simon Guggenheim Memorial Foundation Fellowship to T.W., a David Bruton Jr. Centennial Professorship to T.W., the Fundacão para a Ciência e a Tecnologia (Portugal), and a 2010 Fulbright International Science and Technology Ph.D. Award to M.S.B.
Disclosure Statement
No competing financial interests exist.