
Abstract
Abstract
A chain tree is a data structure for representing changing protein conformations. It enables very fast detection of clashes and free potential energy calculations. The efficiency of chain trees is closely related to the bounding volumes associated with chain tree nodes. A protein subchain associated with a node of a chain tree will clash with another subchain only if their bounding volumes intersect. It is therefore essential that bounding volumes are as tight as possible while intersection tests can be carried out efficiently. We compare the performance of four different types of bounding volumes in connection with the rotation of protein bonds. It is observed that oriented bounding boxes are not as good as could be expected judging by their extensive use in various applications. Both rectangular- and line-swept spheres are shown to have very good tightness of fit but the line-swept, or even simple spheres, are shown to be significantly faster because of quick overlap checks. We also investigate how the performance of the recently introduced adjustable chain trees is affected by different bounding volume types.
1. Introduction
The chain tree is a binary tree in which each node is a subchain of atoms. Each node is, furthermore, associated with a transformation matrix and a bounding volume. To maintain the conformation of a protein chain during folding, a brute-force method can be employed that requires Θ(n) update time for each conformational change and Θ(n2) time to detect clashes. Lotan et al. (2004) showed that when using a chain tree, this is reduced to Θ(log n) update time and Θ(n4/3) clash-detection time.
We previously suggested (Winter and Fonseca, 2011) a modification of chain trees based on the assumption that portions of folding proteins (such as α-helices and β-strands) are formed relatively early in the folding process, and they remain stable throughout many (if not all) iterations of the simulation. Bonds of such subchains can be locked and their bounding volumes therefore remain unchanged. As a consequence, the chain tree can be rearranged (so that locked subchains are tightly covered by a single interior node of the chain tree) and rebalanced. The adjustable chain tree has an improved running time compared to the standard chain tree and the asymptotic running time stays unchanged.
We study four common bounding volumes that are reasonable for clash detection in protein chains. These are oriented bounding boxes, rectangular-swept spheres, line-swept spheres, and spheres (Fig. 1). The computational results indicate that spheres, with their trivial intersection test and simple update methods, perform equally well as line-swept spheres that have a better tightness of fit. Oriented bounding boxes and rectangular-swept spheres are clearly inferior. This is particularly surprising as oriented bounding boxes have been a standard choice for this type of application. As chain trees are adjusted to the protein to fit peptide planes and secondary structures more tightly, line-swept spheres become slightly more efficient than spheres, while oriented bounding boxes and rectangular-swept spheres remain inferior.
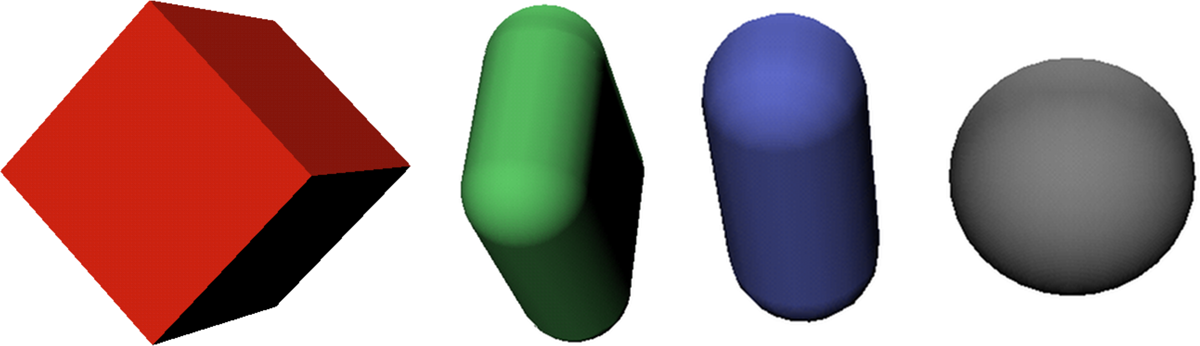
The four bounding volume types compared: Oriented bounding box (OBB), rectangular-swept sphere (RSS), line-segment swept sphere (LSS), and sphere (PSS).
This paper is organized as follows. A brief description of proteins, chain trees, and adjustable chain trees is given in Section 2. The different types of bounding volumes are discussed in Section 3. Computational results are given in Section 4, and, finally, concluding remarks and suggestions for further research are collected in Section 5.
2. Protein Chain Trees
A protein is an organic compound made of n amino acids arranged in a linear chain. A given sequence of amino acids always folds into the same three-dimensional (3-D) structure referred to as the native conformation of that sequence. The lengths and angles between adjacent atoms in the chain are mostly fixed due to steric interactions as shown in Figure 2, so the conformation of the entire protein can be completely defined by the torsions around the covalent bonds.
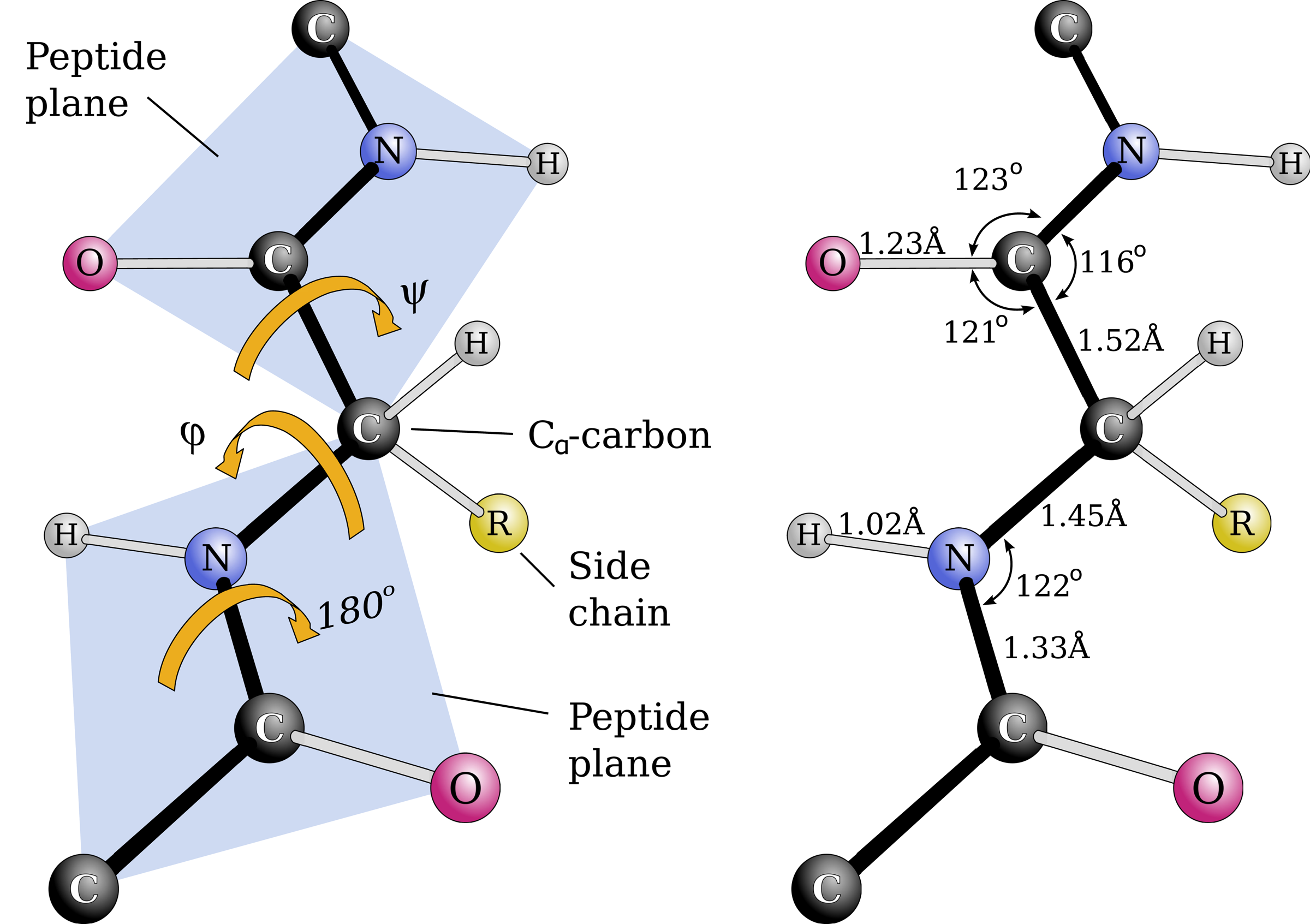
Backbone geometry and definitions. (Left) Since the C-N bond can be fixed at 180°, peptide planes containing six backbone atoms each are formed. The angles around the N-C α and C α -C bonds (Φ, Ψ) are the torsion angles of the chain. (Right) Typical backbone bond lengths and angles.
Protein structure prediction is the problem of predicting the native conformation. This is typically done using Monte Carlo simulations or other metaheuristics, in which a conformation is iteratively improved by changing one or more torsion angles slightly.
After each rotation around a bond, a check is performed to ensure that no two atoms clash. A brute-force method to do this is to first transform all atoms succeding the bond, which takes
The chain tree (Lotan et al., 2004) is a data structure for maintaining the conformation of a chain, which enables
An adjustable chain tree (Winter and Fonseca, 2011) is a modification of the standard chain tree. In adjustable chain trees, bonds in secondary structures, such as α-helices and β-strands, are grouped so that they have the same lowest common ancestor. As a result of this grouping, adjustable chain trees are rebalanced (regarding roots as nodes of height 0), and the bounding volumes are recomputed to tightly fit their underlying atoms (not just their two children). Bounding volumes associated with nodes of secondary structures will be much tighter as these structures fit very well into most bounding volumes.
Adjustable chain trees are also adjusted so that every three bonds of each peptide plane have a common lowest ancestor. This results in very tight bounding volumes at the lowest levels of the chain tree. A detailed description of adjustable chain trees, as well as computational results documenting the speed-up, can be found in Winter and Fonseca (2011).
3. Bounding Volumes
There are four operations that are required to use a particular type of bounding volume in the chain tree:
• transform a bounding volume to another coordinate system, • decide if two bounding volumes intersect, • find a tight volume bounding two smaller volumes, and • find a tight volume bounding a set of bonds or atoms.
Assuming that all backbone atoms have the same radii, the fourth operation can be reduced to finding the minimum volume bounding a set of points. The following subsections discuss how these operations are performed for four different types of bounding volumes: oriented bounding boxes, rectangular swept spheres, line-segment swept spheres, and point-swept spheres.
3.1. Oriented bounding boxes
An oriented bounding box (OBB) is a rectangular block with an arbitrary orientation (Gottschalk et al., 1996). There are many possible representations of OBBs. The center point, an orthonormal set of three orientation vectors and three numbers indicating the extents of the box, have been used in this study.
To transform a point from one coordinate system to another, both a translation and a rotation of the point is necessary. For vectors, however, only the rotation should be performed. The coordinate transformation of an OBB is obtained by transforming (rotating and translating) the center point and rotating the orientation vectors.
To decide if two OBBs, Bl and Br, in 3-D intersect, it is not enough to check if the vertices of Bl are all on the outside of Br, and vice versa. Instead, it is observed that Bl and Br are disjoint if there exists a plane that separates Bl and Br. The projections of Bl and Br onto a line (axis) orthogonal to the separating plane will correspond to two disjoint line-segments if there is no overlap. It can be shown that it is enough to check 15 such axes: 6 corresponding to all orientations of Bl and Br and 9 defined by pairs of orientation vectors, one from Bl and one from Br, respectively (Ericson, 2005).
OBBs bounding a point set can be determined using principal components analysis (PCA) as discussed in Ericson (2005). The three principal component vectors are used as the orientation of the box. The center and extents can then easily be defined so the box is as small as possible given the orientation.
The OBB bounding two smaller OBBs is determined using the above method applied to the 16 corners of a given pair of OBBs. There are several methods to improve the tightness of a bounding OBB, such as using PCA only on the convex hull of the points. We have not implemented these improvements.
3.2. Rectangular swept spheres
A rectangular swept sphere (RSS) is defined as the Minkowski sum of an arbitrarily oriented flat rectangle and a sphere (Larsen et al., 2000; Eberly, 2000). The rectangle is represented by a center point, two orthogonal vectors (defining the orientation), and two numbers specifying the extents of the rectangle. An RSS is transformed into another coordinate system in a similar way as OBBs are.
The intersection test between two RSSs is described in Larsen et al. (2000) and amounts to computing the distance between their defining rectangles Rl and Rr in 3-D. Assume first that the closest pair of points • The open upper half-space bounded by the plane through pli with • The open upper half-space bounded by the plane through prj with
If these conditions are not met for any pair of edges, then either pl or pr are in the interior of one of the rectangles. In this case, the distance between Rl and Rr is the largest of the following two values: The separation between Rl and Rr projected onto an axis normal to Rl or projected onto an axis normal to Rr.
A method for determining an RSS bounding a point set is described in Larsen et al. (2000). First, the principal components,
An RSS bounding two RSSs with radii rl and rr is obtained in the following way. First an RSS bounding the eight corner points of the two rectangles is constructed. Its radius is then extended by max(rl, rr).
3.3. Line-segment swept spheres
A line-segment swept sphere (LSS) is the Minkowski sum of an arbitrarily oriented line-segment and a sphere. An LSS is therefore fully specified by the two end points of the line segment and the radius of the sphere. An LSS is also sometimes referred to as a line-swept sphere, a capsule, a capped cylinder, a spherocylinder (Ericson, 2005), or even as a cigar (Bereg, 2004).
An LSS is transformed to another coordinate system by applying the appropriate transformation matrix to the end points of its defining line segment. Two LSSs intersect if the shortest distance between their defining line segments is shorter than their combined radii. A very fast method to find the distance between two line segments is described in Ericson (2005).
To find a good LSS bounding a set of points, the first principal component,
To find a good LSS, B, bounding two smaller LSSs Bl and Br, the four spheres defined by the end points of the line segments of Bl and Br and their respective radii are considered. The direction
3.4. Point swept spheres
A point-swept sphere (PSS) is simply a sphere. We use the term PSS to emphasize its relation to the other two bounding volumes introduced above and to have a meaningful abbreviation. A PSS is represented by its center and radius. The coordinate transformation is done simply by transforming the center-point.
Two PSSs intersect if the distance between their centers is less than their combined radii. Finding the PSS bounding two PSSs is trivial. (See, e.g., Ericson, 2005, for further details.) Welzl's algorithm (Welzl, 1991) gives an expected linear time algorithm for finding the minimum radius sphere bounding a set of points. Note that, contrary to the other three volume types, the PSS is the only one that guarantees a minimum bounding volume.
4. Computational Results
The improvement in efficiency of the adjustable chain tree over the standard chain tree was documented in Winter and Fonseca, 2011). This article focuses on analyzing which bounding volume gives the best efficiency, both in the standard and the adjustable chain tree. First, the data set used for the different experiments is described. Second, a cost measure is defined, which can be used to evaluate the speed-up independently of implementational and hardware details. Finally we show to what extent different bounding volume types speed up clash-detection.
4.1. Data set
The same data set as in Winter and Fonseca (2011) is used. The first five chains have a similar length of 110–119 residues, the typical length in PDBSelect 25 (Berman et al., 2000). They are selected so that they have different fractions of residues in secondary structures (Table 1). The last four chains are from the seminal paper on chain trees (Lotan et al., 2004). The lengths vary from 68 to 755 residues. Figure 3 shows all nine proteins.
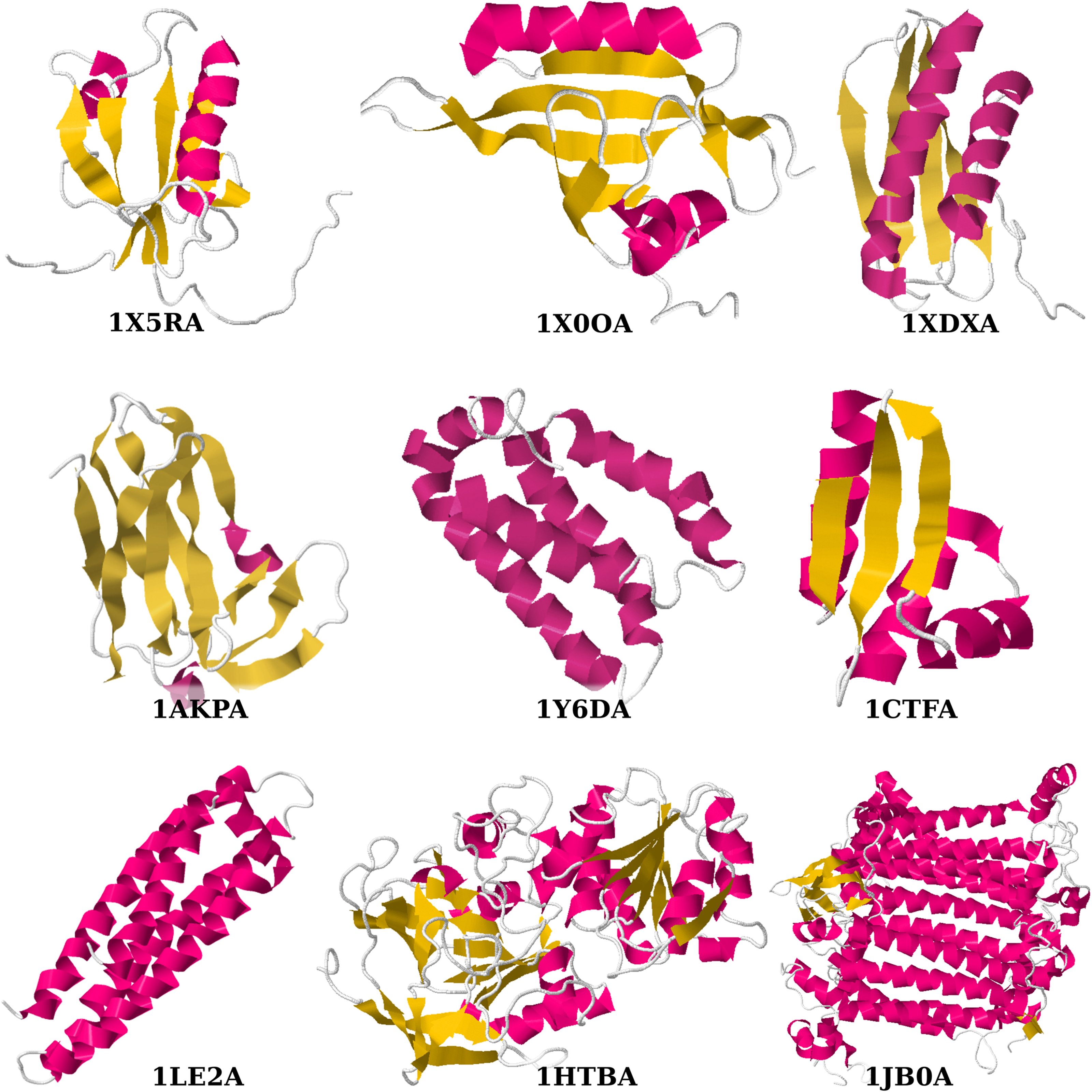
Chains in the two data sets. The first five chains have almost the same length, but different distributions of secondary structures. The last four chains are from Lotan et al. (2004) and have varying lengths.
Second Column is the Number of Residues in the Examined Proteins. The Third Column Indicates the Portion of Residue in Secondary Structures. The Fourth Column Indicates the Number of α-Helices and β-Strands.
4.2. Cost measure
To evaluate the efficiency of a particular bounding volume independent of implementational and hardware issues, we determine the average cost of a single rotation in the chain tree. The cost of a rotation is defined as
where
• NV is the number of bounding volume pairs tested for overlap, • CV is the cost of testing two bounding volumes for overlap, • NU is the number of bounding volumes updated due to rotations, • CU is the cost of updating a bounding volume, • NP is the number of primitive pairs (bonds) tested for overlap, • CP is the cost of testing a primitive pair for overlap.
The three costs, CV, CU, and CP shown in Table 2, were determined experimentally from the chain trees of the native conformations in the data set. A 1° rotation of every nonlocked bond (all peptide bonds and all bonds in secondary structures were locked) was carried out. The subsequent clash detection typically involved many volume intersection tests. CV was determined by measuring the average CPU-time for all volume intersection tests (no matter their outcome). The transformation of one bounding volume into the coordinate system of another was included in CV. Rotations that did not result in clashes were used to update the chain tree. Such an update requires updating all bounding volumes associated with nodes between the rotated bond and the root of the chain tree. CU was determined by measuring the average CPU-time for all such bounding volume updates. The transformation of the bounding volume of the right subtree to the coordinate system of the bounding volume of the left subtree was included in CU. To increase the accuracy of CV and CU, their computation times were averages of 100 repetitions.
The cost of testing a pair of primitives does not depend on the bounding volume type.
Testing a pair of primitives corresponds to finding the distance between two atoms. To determine CP, we therefore generated 106 random point-pairs and measured the average time it took to transform one point to another point's coordinate system and find their distance.
The number of bounding volume pairs tested for overlap, NV, will be a good indicator of a volumes ``tightness of fit.’’ However, to measure the tightness of fit explicitly we introduce the relative volume difference between two volume types. Given an adjustable chain tree representing a protein structure, the relative volume difference between two bounding volume types, BV1 and BV2, is defined as
where the sum is over all nodes (interior and leaves) in the adjustable chain tree (N) and VBV (n) is the volume of the bounding volume associated with node n.
4.3. Comparison of bounding volumes
In all experiments reported in this subsection, peptide bonds were locked in the adjustable chain tree. The Φ torsion angle on the first N-C α bond and the Ψ torsion angle on the last C α -C bond do not affect the conformation of the protein, so the corresponding bonds were therefore also locked in the adjustable chain trees. Bonds in secondary structure segments were also locked.
Two sets of experiments were carried out. In the first set, both standard chain trees and adjustable chain trees were set up to represent native conformations of the selected chains. The coordinates of all atoms were obtained from the Protein Data Bank (PDB) (Berman et al. 2000). For each of the nine chains, 10 rotations by the angles
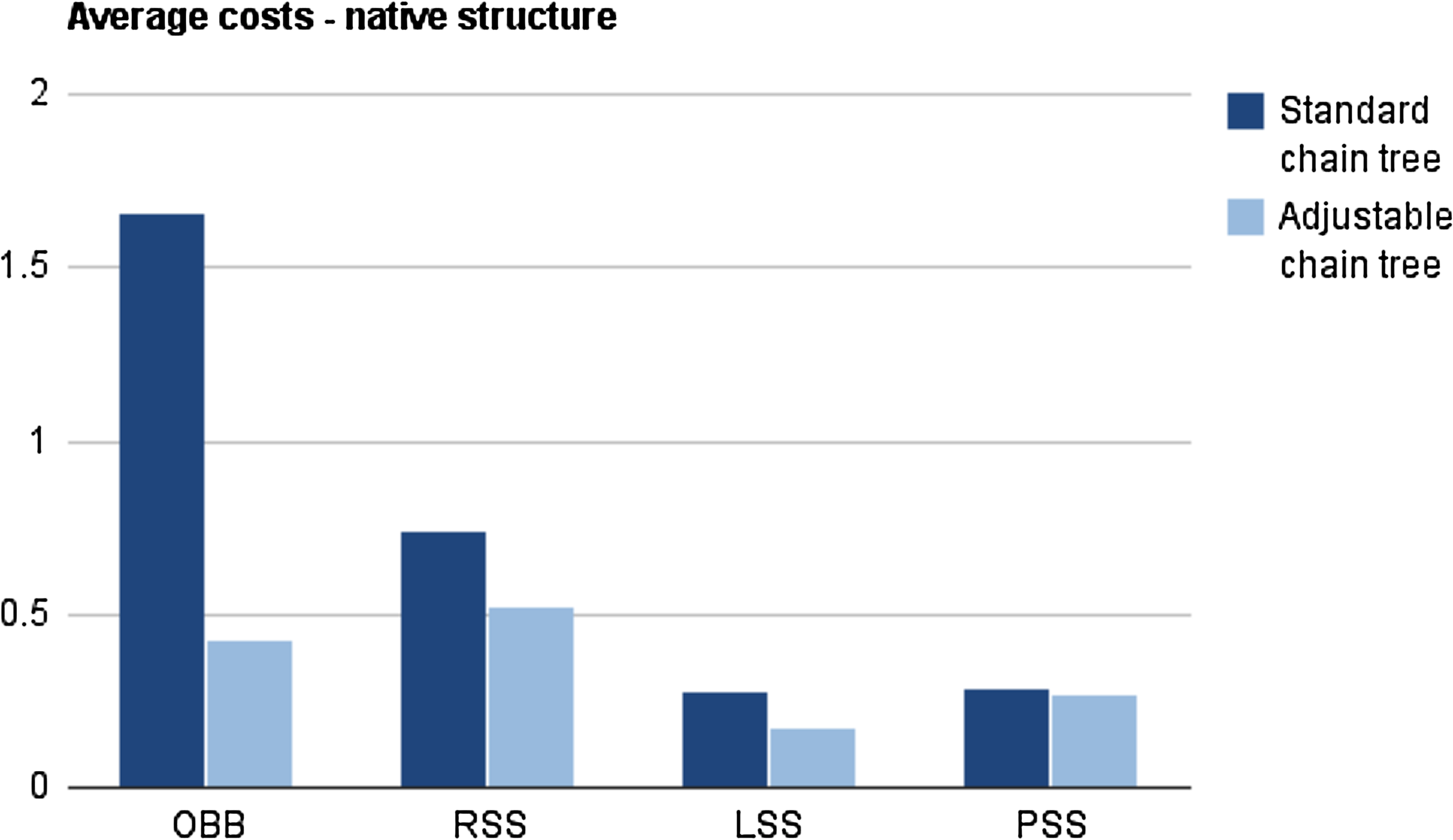
Average costs (in ms) of a rotation in a standard chain tree and an adjustable chain tree. These measurements are performed on the native conformation, which is typically as compact as possible for a protein.
It is clear that the adjustable chain trees with grouped peptide planes and secondary structures provide a substantial speed-up when using both OBBs, RSSs, and LSSs. Surprisingly, this speed-up is not observed for PSSs. The reason for this is that spheres have no elongation. Their tightness of fit, therefore, does not improve when the adjustable chain trees group long segments of α-helix and β-strand. However, for the standard chain trees, spheres are one of the fastest bounding volume types together with LSSs. This indicates that the results in the seminal paper on chain trees (Lotan et al., 2004) could have been improved significantly by replacing the RSSs with LSSs or PSSs.
Another significant feature of Figure 4 is that the average cost of OBBs decreases dramatically when using adjustable chain trees. The reason is that OBBs have sharp corners. Therefore, in the standard chain trees, OBBs of nodes just above leaves are significantly larger than the corresponding swept sphere volumes (Figure 5). In the adjustable chain trees, the four atoms in peptide planes are collected under a single node and the bounding volumes are calculated based on atom positions and not children volumes. This improvement greatly reduces the size of OBBs in particular.

OBBs and LSSs of the two lowest levels in a standard chain tree. The volume of OBBs quickly increases when climbing the tree because they bound the corners of their children.
The cost measure is made up of three terms. For all volume types, the main contribution to the average cost is the term NVCV. For any given rotation, NU is rarely larger than 10 and NP is typically less than 50. As shown in Figure 6, however, NV, for the proteins in our dataset, is typically in the range between 200 to 500, which makes the overlap checks of bounding volumes the most time-consuming part of a rotation. From Figure 6 it is noted that both in terms of NV and relative volume difference (averaged over adjustable chain trees for proteins specified in Section 4.1), the RSSs are the most tight-fitting bounding volumes closely followed by LSSs. Since CV of the RSSs is 3 times that of LSSs, however, the total CVNV term of LSSs is smaller.
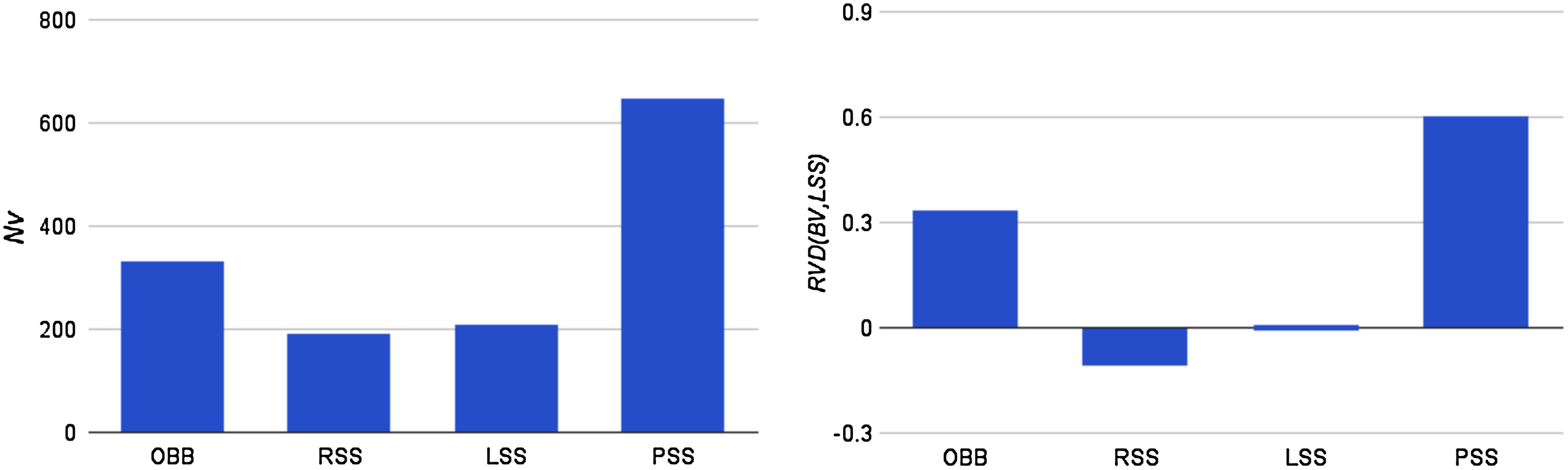
The tightness of fit for different bounding volume types, (left) the average number of overlap checks in a rotation, NV and (right) the relative volume difference between a particular volume type and an LSS.
As mentioned, both LSSs and PSSs perform well in the standard chain trees but LSSs are slightly faster. For adjustable chain trees the LSSs are 37% faster than PSSs, so it is safe to conclude that the tightness of fit that the LSSs have outweigh the fast overlap check of the PSSs. The main conclusion of this paper is, therefore, that LSSs are the optimal choice of bounding volumes for protein chain trees. We assume that this result holds true for bounding volume hierarchies of chains in general.
The native conformations, that are used as starting conformations for the experiments above, are all very tightly packed. To verify that the results hold with a more loosely packed conformation where there are less clashes after each rotation, the experiments above were repeated with partially unfolded conformations. As shown in Figure 7, the same trends as in Figure 4 are observed. To check that the cost measure is not overly simplistic, the average CPU-time of a rotation was also measured. As shown in Figure 8, the same trend is observed again.
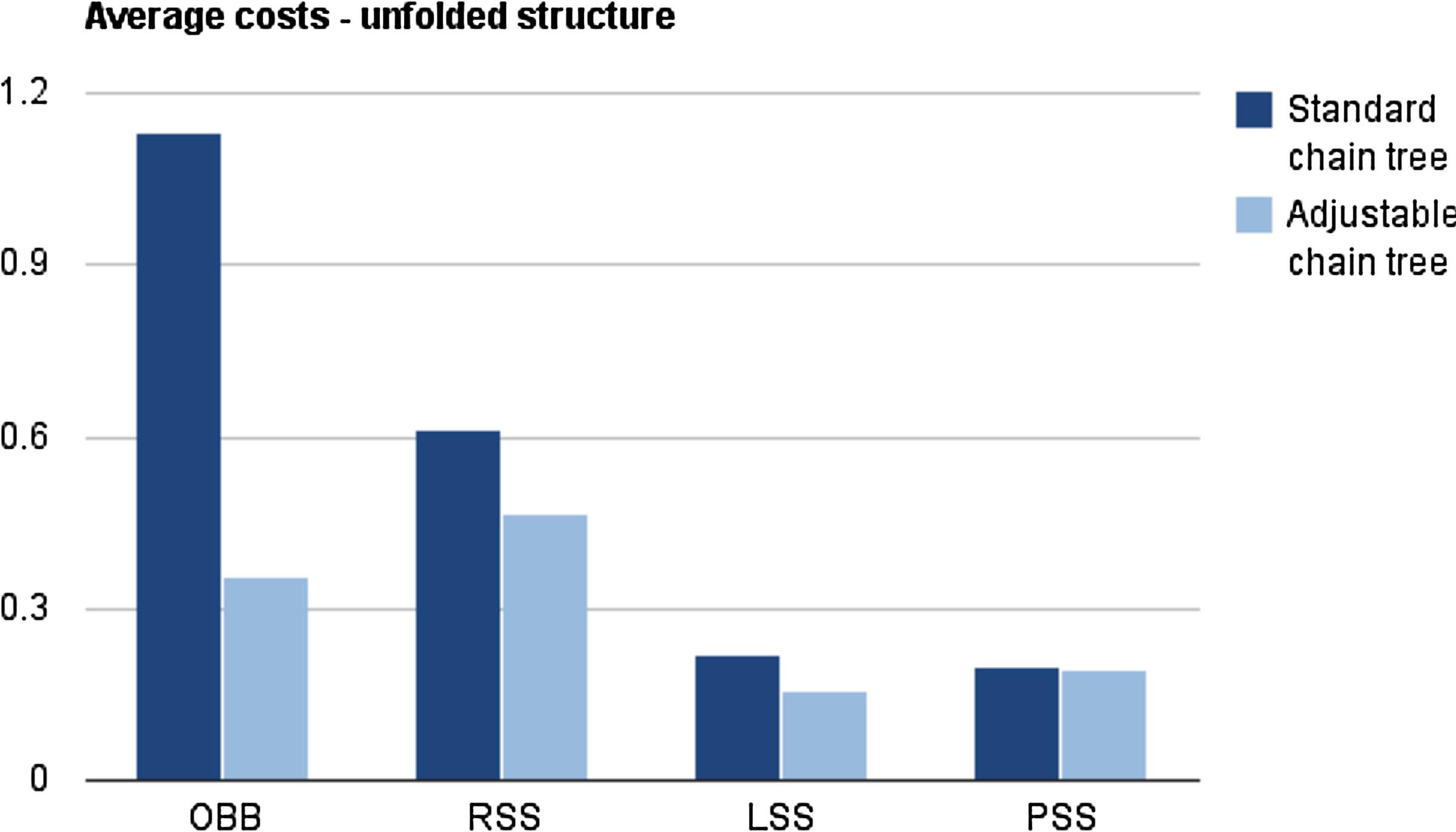
Average costs (in ms) of a rotation in a standard chain tree and an adjustable chain tree. These measurements are performed on a partially unfolded conformation.

Average CPU-time (in ms) of a rotation in a standard chain tree and an adjustable chain tree.
5. Conclusions
In this article we compared different bounding volumes that can be associated with the nodes of chain trees. LSSs seem to perform much better than OBBs and RSSs that have been used as standard bounding volumes in other applications of chain trees to proteins. The performance of bounding volumes was shown to be a tradeoff between fast collision checks and tightness of fit. Our results clearly indicate that RSSs are the most tight-fitting volumes, but a substantial speed-up is possible when using LSSs because of their fast overlap checks. Another, perhaps somewhat surprising conclusion is that, at least for shorter chains, PSSs perform comparable with LSSs.
Bounding volumes also play an essential role when adjustable chain trees are used for the efficient determination of free energy changes when moving from one conformation to another. Bounding volumes will, in this context, typically have greater radius and more intersection tests will be required. This is not only due to the increased volumes but also to the fact that the entire chain tree has to be checked for overlaps. We, therefore, believe that adjustable chain trees and LSSs will prove even more useful in energy calculations than they did in clash-detection.
Footnotes
Disclosure Statement
No competing financial interests exist.
Appendix
| OBB | RSS | OBB | OBB | ||||
|---|---|---|---|---|---|---|---|
| SCT | ACT | SCT | ACT | SCT | ACT | SCT | ACT |
| 0.556 | 0.414 | 0.124 | 0.112 | 0.235 | 0.351 | 0.102 | 0.119 |
| 0.555 | 0.403 | 0.117 | 0.098 | 0.226 | 0.323 | 0.102 | 0.099 |
| 0.495 | 0.354 | 0.097 | 0.081 | 0.175 | 0.260 | 0.079 | 0.091 |
| 0.501 | 0.381 | 0.108 | 0.120 | 0.199 | 0.324 | 0.100 | 0.125 |
| 0.604 | 0.355 | 0.107 | 0.064 | 0.140 | 0.211 | 0.060 | 0.051 |