
Abstract
Abstract
Although de novo motifs can be discovered through mining over-represented sequence patterns, this approach misses some real motifs and generates many false positives. To improve accuracy, one solution is to consider some additional binding features (i.e., position preference and sequence rank preference). This information is usually required from the user. This article presents a de novo motif discovery algorithm called SEME (sampling with expectation maximization for motif elicitation), which uses pure probabilistic mixture model to model the motif's binding features and uses expectation maximization (EM) algorithms to simultaneously learn the sequence motif, position, and sequence rank preferences without asking for any prior knowledge from the user. SEME is both efficient and accurate thanks to two important techniques: the variable motif length extension and importance sampling. Using 75 large-scale synthetic datasets, 32 metazoan compendium benchmark datasets, and 164 chromatin immunoprecipitation sequencing (ChIP-Seq) libraries, we demonstrated the superior performance of SEME over existing programs in finding transcription factor (TF) binding sites. SEME is further applied to a more difficult problem of finding the co-regulated TF (coTF) motifs in 15 ChIP-Seq libraries. It identified significantly more correct coTF motifs and, at the same time, predicted coTF motifs with better matching to the known motifs. Finally, we show that the learned position and sequence rank preferences of each coTF reveals potential interaction mechanisms between the primary TF and the coTF within these sites. Some of these findings were further validated by the ChIP-Seq experiments of the coTFs. The application is available online.
1. Introduction
By only examining the over-representation of sequence patterns, the previous generation motif finders often miss some real motifs and generate many false positives. On the other hand, additional information for the input sequences are found to be helpful for improving motif finding. For example, some transcription factor (TF) binding motifs (e.g., TATA-box) are localized to certain intervals with respect to the transcription start site(s) (TSS) of the gene. In this case, the position information can help to filter spurious sites. In protein-binding microarray (PBM) (Berger and Bulyk, 2006) data, the de Bruijn sequences are ranked by their binding affinities, and we expect the correct motif occurs in the high-ranking sequences; such data has a rank preference. In the chromatin immunoprecipitation sequencing (ChIP-Seq) data (Valouev et al., 2008), the ChIPed TF's motif (ChIPed TF is the TF pulled down in the ChIP experiment) prefers to occur in sequences with high ChIP intensity and also near the ChIP peak summits (thus having both position and rank preference). Hence, if we know the position preference and the sequence rank preference of the TF motifs in the input sequences, we can improve motif finding. In fact, many existing motif finders already utilize such additional information. MDscan (Liu et al., 2002) only considers high-ranking sequences to generate its initial candidate motifs. Other programs allow users to specify the prior distribution of position preference or sequence rank preference (Bailey and Elkan, 1994; Pavesi et al., 2001; Bailey, 2011; Kulakovskiy et al., 2010; Hu et al., 2010) or add such preferences as a prior knowledge component in their scoring functions (Chen et al., 2007; Narang et al., 2010; Linhart et al., 2008; Keilwagen et al., 2011; Frith et al., 2004). However, the users may not know the correct prior(s) to begin with. Even worse, different motifs may have different preferences. For example, in ChIP-Seq experiments, some motifs prefer to occur in high-ranking sequences and at the center of the ChIP peak summit while others do not.
To resolve such problems, we propose a novel motif-finding algorithm called SEME (sampling with expectation maximization for motif elicitation). SEME assumes the set of input sequences is a mixture of two models: a motif model and a background model. It uses an EM-based algorithm to learn the motif pattern (PWM), position preference, and sequence rank preference at the same time, instead of asking users to provide them as inputs. SEME does not assume the presence of both preferences but automatically detects them during the motif refinement process through statistical significance testing. We also observe that EM algorithms are generally slow in analyzing large-scale, high-throughput data. Speeding up EM using suffix tree was recently proposed (Reid and Wernisch, 2011), but the technique cannot be applied when one wants to also learn the position and sequence rank preferences. To improve the efficiency, SEME developed two EM procedures. The two EM procedures are based on the observations that the correct motifs usually have a short conserved pattern in it and majority of the sites in the input sequences are non-motif sites. The first EM procedure, called extending EM (EEM), starts by finding all over-represented short l-mers and then attempts to include and refine the flanking positions around the l-mers within the EM iterations. This way, SEME recovers the proper motif length within a single run, thus saving a substantial amount of time by avoiding multiple runs with different motif length, as done in many existing motif finders (Bailey and Elkan, 1994; Pavesi et al., 2001; Linhart et al., 2008; Kulakovskiy et al., 2010; Hu et al., 2010). The second EM procedure, called the resampling EM (REM), tries to further refine the motif produced by EEM. It is based on a theorem similar to importance sampling (Glynn and Iglehart, 1989), which stated that the motif parameters can be learned unbiasedly using a biased subsampling. By this principle, we can sample more sites that are similar to the EEM's motif and less sites from the background. This way, REM is able to learn the correct motifs using significantly less background sites. In our implementation, REM is capable of producing the correct TF motifs using approximately 1% of the sites normally considered in a typical EM procedure.
Using 75 large-scale synthetic datasets, we show that SEME is better both in terms of accuracy and running time when compared to MEME (Multiple EM for Motif Elicitation), a popular EM-based motif-finding program (Bailey and Elkan, 1994). We found that MEME is unable to find motifs with gap regions while SEME's EEM procedure can successfully extend the motifs to include them. In the real experimental datasets, we perform comparisons using 32 metazoan compendium datasets and 164 ChIP-Seq libraries. SEME consistently outperformed seven existing motif-finding programs that we compared. In general, we found that SEME not only finds more TF motifs but also gives more accurate results, as evaluated using either PWM divergence, AUC score, or STAMP's p-value (Mahony et al., 2007). Other TFs that bind nearby and function together with the ChIPed TF are called coTF. When we compare the programs to find coTF motifs from 15 ChIP-Seq datasets, the superior performance of SEME is more pronounced. We propose that SEME's ability to learn the underlying motif-binding preference is crucial in its performance. We further confirmed the correctness of the position and sequence-rank preference of the coTF motifs learned by SEME on three ChIP-Seq datasets. The actual ChIP-Seq data of the predicted coTFs clearly shows that SEME managed to infer the correct preferences. We also show that such preferences provide biological insights to the mechanism of the ChIPed TF–coTF interactions.
2. Seme Algorithm
SEME uses a probabilistic framework known as the two-component mixture model (TCM), which is first proposed by MEME (Bailey and Elkan, 1994). It assumes that the observed data is generated by two independent components: a motif model and a background model. Given an ordered list X of equal-length DNA sequences, each site Xi in X is associated with a DNA sequence
We use a naive Bayesian approach to combine three types of preferences (sequence, position, rank):
For sequence preference, we model the motif-site sequence with a position weight matrix (PWM) Θ, and the background sequence with a 0-order Markov model θ0. Θ is a 4×w matrix where Θj,a is the probability that the nucleotide a occurs at position j. For any length-w sequence Xi, the probability that Xi is generated from the motif model and the background model are as follows.
where
The position and sequence-rank preferences are modeled using multinomial distributions. The position preference models the preference of the motif site to certain positions. Similarly, the sequence rank preference tries to model if the motif site prefers the sequences with certain range of ranks, assuming input sequences are ordered by some criteria. To this end, we discretize both the positions and sequence ranks into K bins. The probability a binding site occurs at the k-th position bin is denoted as αk, for
Similarly, for sequence-rank preferences, the probability a motif site occurs at the k-th sequence rank bin is denoted as βk and,
Let Pr(Zi = 1) be λ. The parameters of the mixture model in SEME are
In this work, we developed four phases in the SEME pipeline (Fig. 1). To search for a good starting point, SEME first enumerates a set of over-represented short l-mers (phase 1) and extends each short l-mer to a proper length PWM motif by the EEM procedure (phase 2). The PWM reported by the EEM procedure will approximate the true motif when its starting l-mer captures the conserved region of the motif. To further refine EEM's PWM motif, SEME applies the resampling EM procedure (phase 3). It is an importance sampling version of the classical EM algorithm that greatly sped up the EM iterations. Finally, the refined PWM motifs are scored and filtered for redundancies (phase 4). Below, we briefly describe these four phases (see Supplementary Section 3, Supplementary Material available online at www.liebertonline.com/cmb).

Algorithm description for SEME Pipeline. AUC; area under the ROC curve; EM, expectation maximization; PWM, position-weighted matrices; REM, resampling EM; SEME, sampling with expectation maximization for motif elicitation.

Pseudocode for extending EM procedure.
In each iteration of the M-step, the EEM procedure will also try to include one additional column into Θ(t) if such extension improves the likelihood. Precisely, for each position
where J is any probability distribution over the nucleotides {A,C,G,T}.
While the length of Θ(t) is less than w, we extend the PWM Θ(t) to include position j, which brings the largest G(j). To avoid over-fitting, the selected column also has to be tested (Chi-square) significantly different from the background frequency θ0. The EEM procedure ends when PWM Θ converges. Finally, the columns in Θ representing the l-mer q will be further diluted (by setting all [1.0,0.0,0.0,0.0] columns representing “A” to [
Let Q(·) be the sampler function, where Q(Xi) = 1 if the sequence Xi is sampled, and 0 otherwise. In Supplementary Theorem 1 (Supplementary Section 3.4), we show that the log likelihood function log Pr(X, Z|Φ) can be unbiasedly approximated by
where each sampled site is weighted by factor
Although Equation 6 is true for any arbitrary sampler function Q(·), running EM using different Q(.) yields different sampling efficiencies. For example, we can use a uniform random sampler, i.e., Pr(Q(Xi)=1)=μ for every
The position and sequence-rank preferences are assumed to be nonexistent at the beginning of the REM iterations (i.e.,

Pseudocode for the resampling EM procedure.
3. Results
3.1. Profiling two novel EM procedures
For each dataset, we ran SEME (EEM only), SEME (EEM+REM), and MEME (the classical EM-based motif finder) and obtained the top five predicted PWMs from each program. To test the goodness of the predicted PWMs, we compared the PWM divergence between the predicted PWMs and the actual planted PWMs. We also generated independent testing sequences with length 400 bp (1000 positive sequences with one implanted motif site, 1000 negative sequences without motif site), and computed the ROC-AUC value for each predicted PWM. Figure 4a shows the comparison result. As expected, the random PWMs have the worst AUC values, while the actual planted PWMs have the best AUC values. EEM's predicted PWMs have significantly better discriminative capability (AUC) and similarity (less PWM divergence) to the actual planted PWM as compared to random PWMs. This indicates that EEM's PWMs are good starting points for the subsequent REM procedure. REM's predicted PWMs further improve the AUC score and are similar to the actual planted PWM (as indicated by the small PWM divergence).
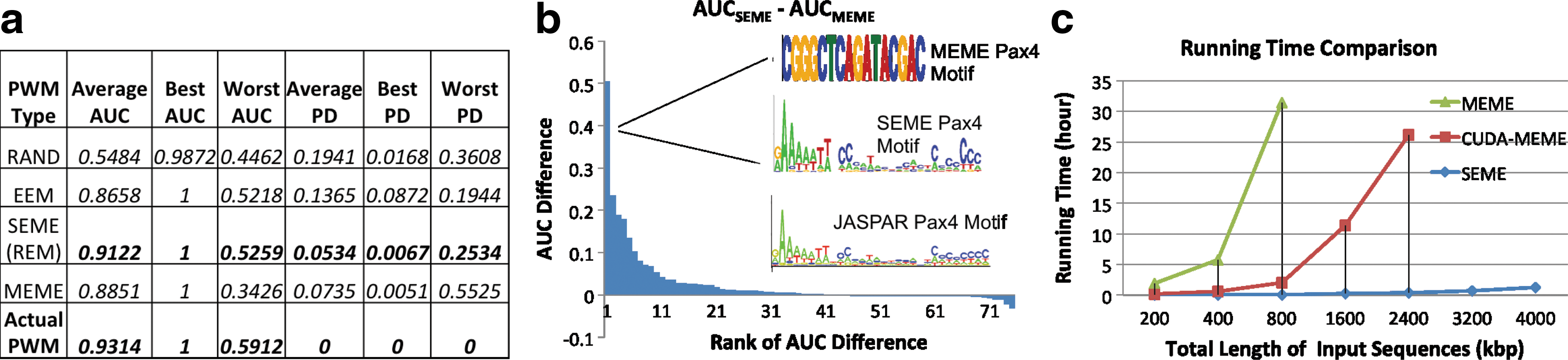
The empirical performance of SEME on synthetic datasets. (
Figure 4a also shows that SEME outperforms MEME. In fact, SEME is better than MEME in 42 out of 75 experiments (the cases with positive AUC differences in Fig. 4b). The cases in which SEME performed worse have relatively small AUC score differences (less than 0.04). We examined the Pax4 dataset in which SEME gains the highest improvement against MEME. The implanted JASPAR Pax4 motif is a diverged PWM of length 30. SEME successfully extended and recovered the full Pax4 motif, thanks to the ability of its EEM procedure to handle long gaps in its extension step. In contrast, MEME failed to model the long gaps due to their starting-point finding procedure, which assumes that all of the PWM positions are equally important.
3.2. Comparing TF motif finding in large-scale real datasets
We compared the performance of SEME with other existing motif-finding programs on two large-scale TF-binding site data. We also study the ability of SEME to uncover the hidden position and/or sequence-rank preferences in the input dataset when they are present.
The result of this comparison is shown in Figure 5a. We find that SEME successfully detected the correct motifs in 21 datasets, whereas the second-best program, Amadeus, succeeded in 18. Weeder and Trawler found correct PWMs in 11 and 12 datasets, respectively. SEME also found more accurate motifs than the rest; it found 12 motifs with PWM divergence <0.12. SEME further detected a significant position preference for the correct motifs for many datasets in this benchmark: most of them tend to bind nearer to the TSS position (see Supplementary Section 1.4 for details).
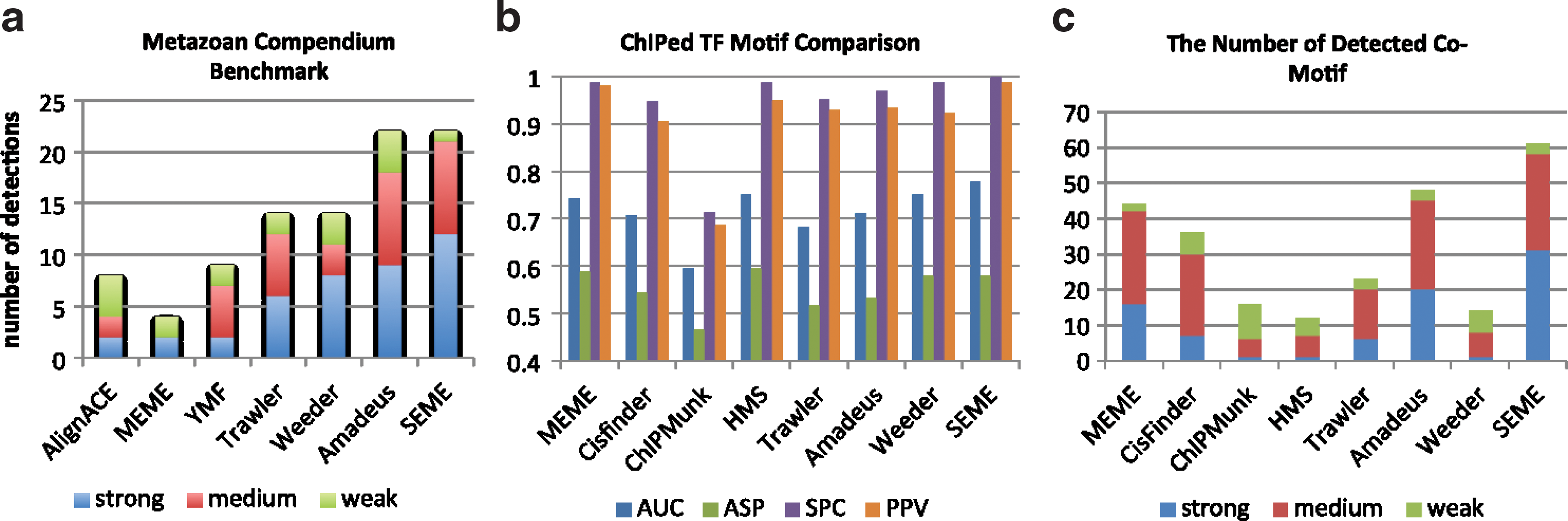
The performance of SEME compared to existing motif-finding programs from large-scale real data. (
We compared SEME with seven popular de novo motif-finding programs for ChIP data: MEME, Weeder, Cisfinder, Trawler, Amadeus, ChIPMunk, and HMS. Each program's top five motifs are evaluated using the four statistics measurements on the test data. For each scoring, the best of the five motifs will be used to represent the performance of a program. Figure 5b shows the average performances of the motif finders. Again, we find that SEME is consistently better than all other programs (first rank in area under ROC curve and positive predictive value and specificity, and third rank in average site performance).
For coTF motif comparison, we used 15 ChIP-Seq libraries whose coTFs have been characterized (the list of coTFs for each ChIP-Seq is in Supplementary Section 2.5). We extracted 400 bp sequences around the ChIP-Seq peaks and compared the top 20 de novo motifs of each program to the known coTF motifs in the JASPAR and Transfac database; we cannot use the previous statistical measurements since coTFs may not occur in all ChIP-Seq peaks. Furthermore, the ChIPed TF binding sites need to be masked before we start the coTF motif finding. SEME and ChIPMunk can do this automatically and, for other programs without auto-masking mode, the input sequences were masked by the top two motifs reported from their ChIPed motif-finding results.
STAMP program (Mahony et al., 2007) was used to compute the p-value of the match between a predicted coTF motif against the known coTF motif. STAMP p-value provides a better match measurement compared to PWM divergence since it removes the motif-length bias (Mahony et al., 2007). We separated the p-value of the PWM matching into three significance levels: (1) weak match (0.05≥p-value>0.01), (2) medium match (0.01≥p-value>0.0001) and (3) strong match (p-value≤0.0001). Figure 5c shows the performances of different motif-finding programs for finding the coTF motifs from the 15 datasets. SEME recovered 61 known coTF motifs, compared to Amadeus and MEME, which find 48 and 44 coTF motifs, respectively. Thirty-one out of the 61 coTF motifs of SEME belong to the strong-match category (Amadeus only found 20) and another 27 are in the medium-match category. This indicates that SEME's predicted coTF PWMs are highly accurate.
To study the biological significance of the learnt preferences, we further studied the output of three datasets, involving the estrogen receptor (ER), androgen receptor (AR), FoxA1, Oct4, and c-Myc TFs, in detail (Fig. 6). The real binding site of each TF was defined to be the site around +/−100 bp around the TF's ChIP-Seq peak, whose known PWM score is better than a cutoff that yields FDR = 0.01. If multiple matches occur, only the best scoring site was chosen. Comparison between SEME's learnt distributions (Fig. 6, middle columns) and the real binding-site distributions (Fig. 6, right-most columns) indicates that SEME is able to learn the correct coTF position and sequence-rank preferences. We also found that the motif positions of FoxA1, a known coTF of ER, is not enriched exactly at the ER ChIP-Seq peak in the MCF7 data; instead it is found in the flanking regions near the ER peaks. Interestingly, in the LnCAP AR ChIP-Seq dataset (FoxA1 is also a known coTF of AR), we found that FoxA1 binds very closely to AR—it is enriched at the AR ChIP-Seq peak summits. This observation is consistent with the previous report that FoxA1 can physically interact with AR (Gao et al., 2003). This observation also indicates the different roles FoxA1 assumes when working with AR and ER (Sahu et al., 2011). In the ChIP-Seq data of Oct4 from mouse ES cells SEME found the motif of c-Myc enriched within Oct4's low-intensity peak regions. We conjectured that, in these regions, Oct4 indirectly bind the DNA through c-Myc (hence explaining the ChIP-Seq's low intensity). An earlier report showed that Oct4, along with Sox2, Nanog, and Stat3, form an enhancer module while c-Myc, along with n-Myc, E2F1, and Zfx, form a promoter module in the ES cell (Chen et al., 2008). In fact, interaction between these enhancer and promotor modules has also been reported previously (Wu and Ng, 2011).
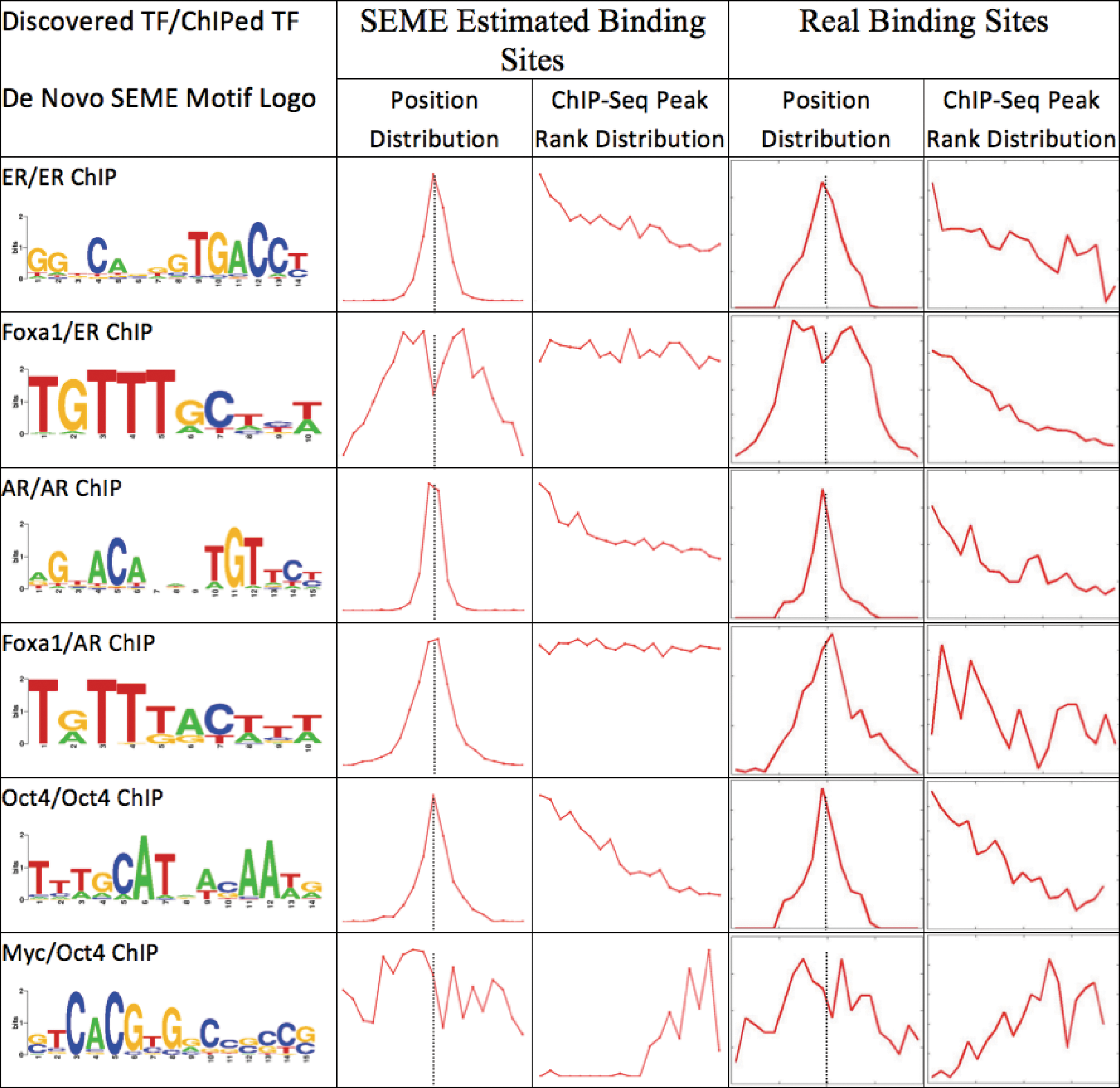
Automatic learning of the position and sequence rank preference from the input data. Instead of requiring the user to input the expected coTF motif preference distribution (position and/or sequence-rank distribution), SEME learns such distributions directly from the input data. We show that most of the time, SEME can learn the correct distributions of each TF (as compared to real binding sites distribution in the rightmost column, defined by the ChIP-Seq and the known PWM of the TF). For position distribution, the x-axis is +/−200 bp from ChIP-seq peak summit (the black dashed line), and the y-axis is the fraction of binding sites in a given position. For rank distribution, the x-axis is the rank of ChIP-seq peak (left: high ChIP intensity, right: low ChIP intensity), and the y-axis is the fraction of binding sites in a given rank. The ChIP-seq peak rank distributions (MCF7 ER ChIP, LNCaP AR ChIP) of FoxA1, and the position distribution of Myc, are tested to be insignificant by SEME.
These examples indicate that the position and sequence-rank distribution learnt by SEME are reasonably accurate, and users could use them to infer the nature of the interaction between the ChIPed TF and the coTF(s). In this manner, SEME can be used to generate biological hypothesis for further experimental validations. Moreover, the highly diverse preferences that we observe highlight the difficulty for users to provide the correct prior in the first place.
4. Conclusion
This article developed a novel algorithm called SEME for mining motifs using mixture model and EM algorithm. We presented three important contributions: (1) automatic detection and learning of the position and sequence-rank preferences of a candidate motif; (2) ability to estimate the correct TF motif length (with possible gaps within); and (3) using importance sampling for efficiency while still able to estimate the EM parameters unbiasedly. As a result, we showed that SEME is substantially better, both in terms of accuracy and efficiency, compared to the existing motif-finding programs.
Moreover, in the task of finding coTF motif in the ChIP-Seq data, SEME not only reports more accurate coTF motifs than other programs but also correctly estimates the position and sequence rank distribution of each coTF's motif. We showed that such information provides useful insights on the interaction between the ChIPed TF and the predicted coTFs. SEME does have a few limitations. Firstly, it assumes that the target motif contains conserved 5-mer region. In cases without such 5-mer, SEME also allows users to provide custom seeds. Secondly, SEME is more suitable for large-scale input (≥100 sequences) since it needs enough samples to determine whether we should do extension (EEM) or include additional binding preferences (REM).
Footnotes
Acknowledgments
This work was supported in part by the MOEs AcRF Tier 2 funding R-252-000-444-112. Z.Z. Zhang is supported by the National University of Singapore research scholarship. C.W. Chang is supported by an A*STAR graduate scholarship.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.