
Abstract
Abstract
Molecular simulation techniques are increasingly being used to study biomolecular systems at an atomic level. Such simulations rely on empirical force fields to represent the intermolecular interactions. There are many different force fields available—each based on a different set of assumptions and thus requiring different parametrization procedures. Recently, efforts have been made to fully automate the assignment of force-field parameters, including atomic partial charges, for novel molecules. In this work, we focus on a problem arising in the automated parametrization of molecules for use in combination with the
1. Introduction
Typically, the number of atoms in biomolecular systems are in the range of 104 to 106. To observe relevant biological phenomena, time scales in the order of nano- to milliseconds need to be simulated. For such large-scale systems, evaluating all atom–atom interactions is practically infeasible. One way of dealing with this is to only consider interactions of atoms whose distance is within a pre-specified cutoff radius. Since not all interactions are considered, an error is introduced. The magnitude of the error due to omitting atom–atom interactions is inversely proportional to the distance between the atoms. More problematically, there are discontinuities as atoms move in and out of the cutoff radius.
Errors and discontinuities are reduced by combining atoms into charge groups, for which individual centers of geometry are determined. If the distance between two centers of geometry lies within the cutoff distance then all interactions between the atoms of the involved charge groups are considered. Ideally, charge groups should be neutral as interactions are then reduced to dipole–dipole interactions that scale inversely proportional to the cubed interatomic distance. Charge groups should not be too large. This is because the effective cutoff distance of an individual atom in a given charge group is given by the cutoff distance minus the distance to the center of geometry of the charge group. If the distance of an atom to the center of geometry becomes large, the effective cutoff becomes small, leading to errors and discontinuities as described above. For the same reason, charge groups should be connected as interatomic bonds impose spatial proximity.
To simulate a molecule, a force field requires a specific topology, which includes the atom types, bonds and angles, the atomic charges, and the charge group assignment. Most biomolecular force fields come with a set of topologies for frequently simulated molecules such as amino acids, lipids, nucleotides, and cofactors. Unparametrized molecules, however, require the construction of their topologies. Such a situation occurs, for instance, when assessing the binding affinity of a novel druglike compound to a certain protein.
Manually building topologies for new compounds is a tedious and time-consuming task, especially when a large chemical library needs to be screened, for example, when determining binding affinities for large sets of potential drug compounds to a newly discovered protein target. Therefore, automated approaches are needed.
Here, we focus on the
As existing automated procedures such as
2. Problem Statement and Complexity
In this section, we give a formal definition of the problem associated with assigning appropriate charge groups within a molecule. Our aim is to capture the two important aspects of chemical intuition discussed above: (1) The number of atoms in a charge group should not exceed a given integer k and (2) the sum of partial and formal charges of a charge group is ideally equal. Mathematically, the latter condition is equivalent to requiring the sum of differences of formal and partial charges in a charge group to be close to zero. We prove
A molecular structure can be modeled as a degree-bounded graph G = (V, E), where the nodes correspond to atoms and the edges to chemical bonds. In addition, we consider node weights
Definition 1
(Charge group partitioning,
Each subset
Theorem 1
Proof
Clearly, the problem belongs to
Definition 2
(Planar three-dimensional matching,
This problem has been shown as
Reduction from
Given a feasible partition to the
We show that there exists a perfect matching if and only if G admits a partition with total error
Suppose
Now assume that no perfect matching exists. First, note that in any optimal solution to the
we get
and the cost contributed to Equation (1) by type 1, 2, and 3 groups becomes equal to mε + c(3 + ε). Together with the remaining n − m − c groups of type 0, the total weight becomes
Using the same reduction but extending the tails to length k − 1 paths with ε weight on the internal vertices and − (k−1)ε weight on the leaf proves the problem to be hard for any k ≥ 4.
For k = 3 and for general, non-planar graphs,
3. Dynamic Programming for Bounded Treewidth
While problem
Although the structural formula of biomolecules is not always a tree, as we will see later, it is usually still treelike, which has already been exploited in Dehof et al. (2011). Formally, this property is captured by the treewidth of a graph (Robertson and Seymour, 1986). The definition is as follows.
Definition 3
A tree decomposition (T, X) of a graph G = (V, E) consists of a tree T and sets Xi for all 1. Every vertex in G is associated with at least one node in 2. For every edge 3. The nodes in T associated with any vertex in G define a subtree of T.
The width of a tree decomposition is max i |Xi| − 1. The treewidth of G is the minimum width of any tree decomposition of G.
In this section, we propose a tree decomposition-based dynamic program for problem
Let (T, X) be a tree decomposition of width ℓ for graph G = (V, E). The high-level idea of the algorithm is as follows (Fig. 2). For an arbitrarily chosen root i of the tree decomposition, we guess the groups that intersect Xi, denoted by the dashed lines in the figure. After removing these groups, G falls apart into connected components, denoted by the filled regions in the figure. By the properties of a tree decomposition, these connected components will correspond one-to-one to the subtrees of the tree decomposition obtained by removing bags that became empty. Recursing on the roots of these new subtrees yields the overall optimal solution.
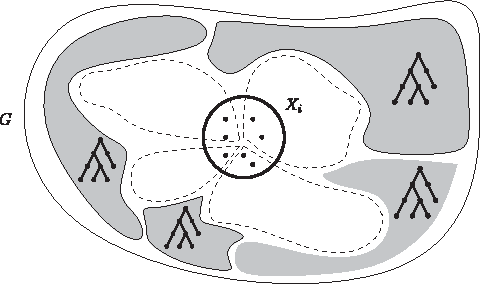
Illustration of the tree decomposition-based dynamic programming algorithm. A graph G falls apart into connected components (gray regions) by removing the groups (dashed lines) that intersect bag Xi.
Without loss of generality we assume that T has at most n := |V | vertices and depth
Definition 4
For vertex sets V1 ⊆ V2 ⊆ V, set
Furthermore, by r(S) we denote the root of a subtree S of T, and for any node i in T and any vertex set A ⊆ V we denote by
which also holds in the base case where
Theorem 2
The cost of an optimal solution to
Proof
Let (T, X) be a tree decomposition of G of width k and depth
Indeed, for each partition
Since every
Consider node (i, A) in
Additionally storing, along with each entry cgp(i, A), the partition
4. Experimental Evaluation
We implemented the dynamic programming method for bounded treewidth in C++ using the
4.1. Hydration-free energy of amino acid side chains
We tested the quality of charge group assignments by comparing the calculated free energies of solvation in water of a set of 14 charge-neutral amino acid side chain analogs to experimental values, which are denoted by ΔGhyd,exp (Gerber, 1998; Oostenbrink et al., 2004). For each analog, we used the
Using the
As described in the introduction, neutral charge groups lead to more accurate simulation results. In our problem definition, we aim to identify a charge group assignment in which the constituent charge groups have small residual error, which is the absolute difference between the sum of the formal charges and the sum of the partial charges of the atoms in the charge group. To ensure neutral charge groups where possible, we adjust the partial charges slightly by redistributing the residual error of every charge group over its atoms.
The results are presented in Table 1 and Figure 3. The
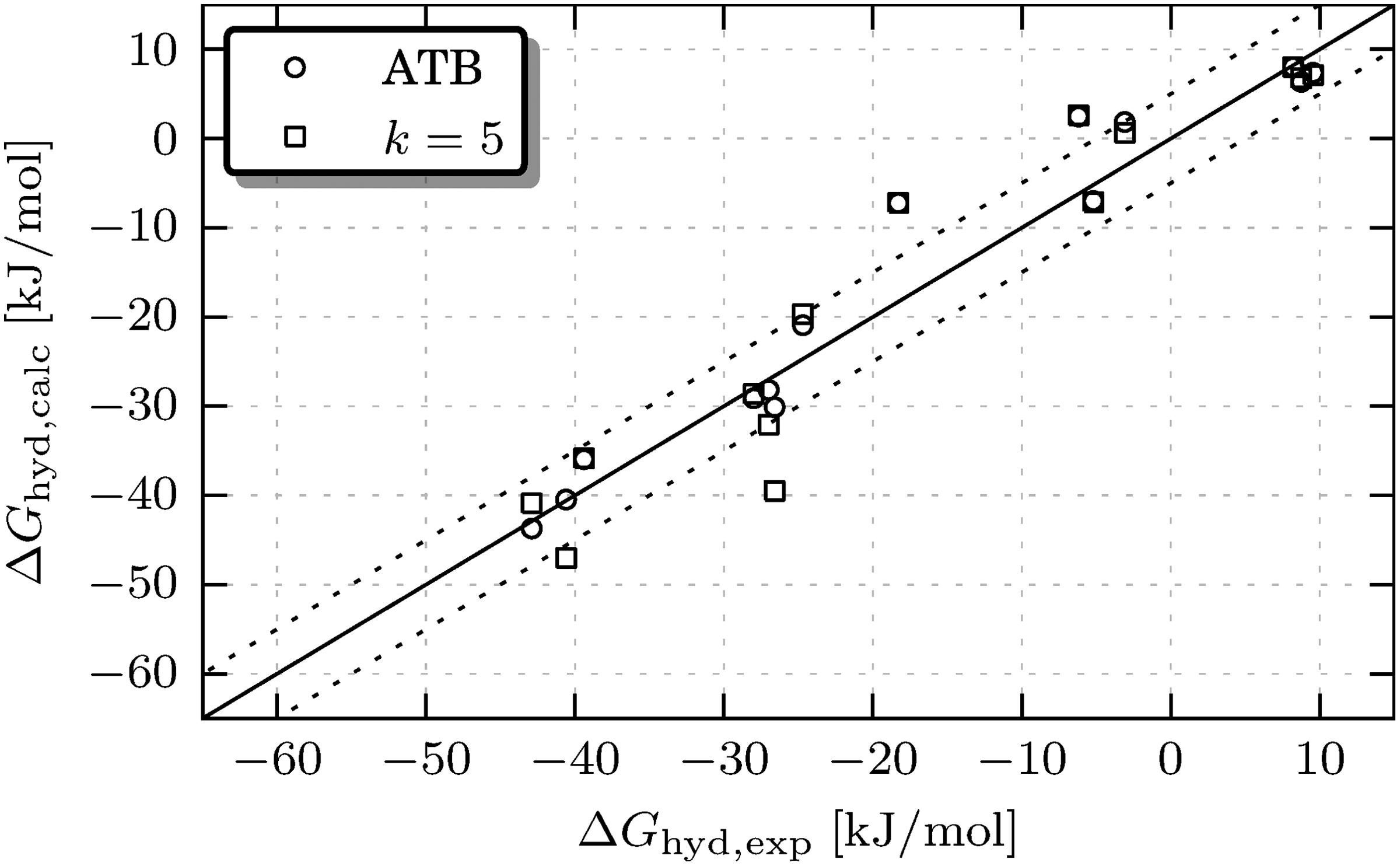
Calculated ΔGhyd values versus experimental ones, showing the effect of the charge group assignment on the simulated hydration-free energy. The labels in the legend are the same as in Table 1. The solid line represents perfect agreement with experiment, dotted lines indicate the ±5 kJ/mol approximate experimental error.
All free-energy values are given in kJ/mol. When two values separated by a semicolon are given, two experimental values were found. The absolute free-energy differences between simulation outcomes and the experimental values are given in parentheses. The average values of these differences are given in the bottom line. “ffG53A6” denotes results using the default
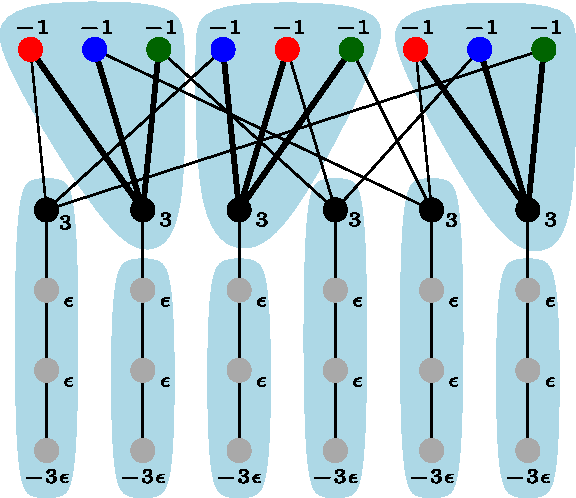
4.2. Adenosine tri-phosphate
Although showing good performance on the amino acid side chains, the
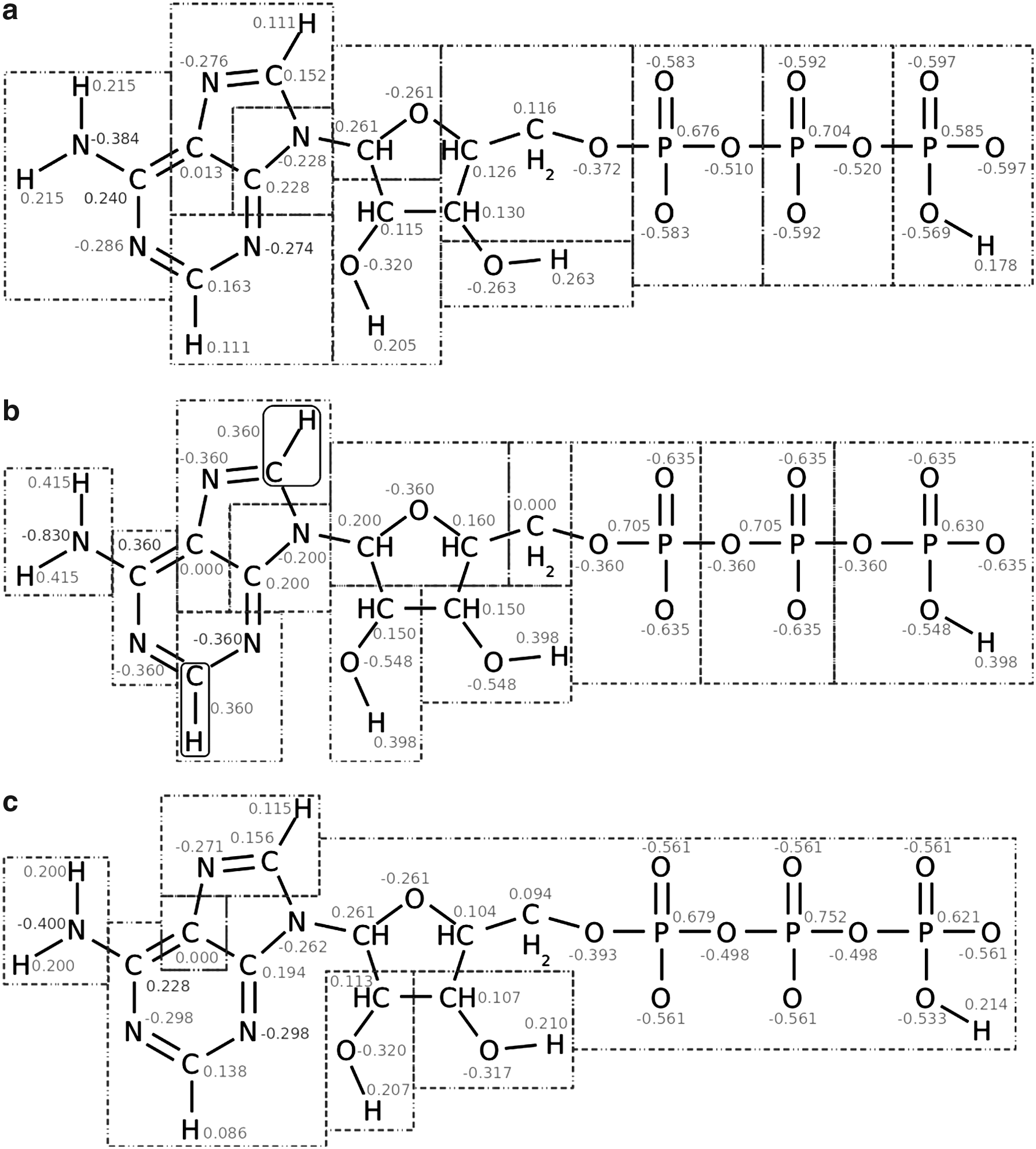
Charge group assignments for adenosine tri-phosphate (ATP) at pH 5.0. The total molecular charge is −3. The partial charges are shown in gray. (
5. Discussion
In this work, we have formally introduced the charge group partitioning problem that arises in the development of atomic force fields, and more generally, in the identification of functional groups in molecules. The problem is to assign atoms to charge groups of size at most k and such that for every charge group the sum of its partial charges is close to the sum of its formal charges. We showed
Algorithmically, we showed that the case k = 2 is solvable in polynomial time. In addition, we have presented a polynomial-time algorithm for bounded charge group size in cases where the molecular graph is a tree. Based on the observation that molecular graphs have bounded treewidth in practice and exploiting further properties such as outerplanarity and bounded degree, we developed a practical dynamic programming algorithm, which is based on a tree decomposition of the graph corresponding to the chemical structure of interest. An interesting open question is to settle the complexity status for the case k = 3.
Since our method relies on point charges obtained from quantum mechanical calculations, the quality of charge group assignments and subsequently of simulation outcomes depends on the accuracy of these calculations. However, our experiments have shown that taking into account charge group size and neutrality already gives good results, especially for large highly charged molecules such as ATP, where other methods fail to produce meaningful solutions. Still, the greedy partitioning algorithm built into the
Footnotes
Acknowledgments
We thank SARA Computing and Networking Services (![]() ) for their support in using the Lisa Compute Cluster. In addition, we are grateful to the referees for helpful comments. The research leading to these results has received support from the Tinbergen Institute as well as from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115002 (eTOX), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/20072013) and EFPIA companies in-kind contribution.
) for their support in using the Lisa Compute Cluster. In addition, we are grateful to the referees for helpful comments. The research leading to these results has received support from the Tinbergen Institute as well as from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115002 (eTOX), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/20072013) and EFPIA companies in-kind contribution.
Author Disclosure Statement
The authors declare that no competing financial interests exist.