Protein interactions are central to all the biological processes and structural scaffolds in living organisms, because they orchestrate a number of cellular processes such as metabolic pathways and immunological recognition. Several high-throughput methods, for example, yeast two-hybrid system and mass spectrometry method, can help determine protein interactions, which, however, suffer from high false-positive rates. Moreover, many protein interactions predicted by one method are not supported by another. Therefore, computational methods are necessary and crucial to complete the interactome expeditiously. In this work, we formulate the problem of predicting protein interactions from a new mathematical perspective—sparse matrix completion, and propose a novel nonnegative matrix factorization (NMF)-based matrix completion approach to predict new protein interactions from existing protein interaction networks. Through using manifold regularization, we further develop our method to integrate different biological data sources, such as protein sequences, gene expressions, protein structure information, etc. Extensive experimental results on four species, Saccharomyces cerevisiae, Drosophila melanogaster, Homo sapiens, and Caenorhabditis elegans, have shown that our new methods outperform related state-of-the-art protein interaction prediction methods.
1. Introduction
Proteins play an essential role in nearly all cellular functions, such as promoting biochemical reactions and composing cellular structures. The multiplicity of functions that proteins execute in most cellular processes and biochemical events is attributed to their interactions with other proteins. As a result, it is critical to understand protein–protein interactions (PPIs) in both scientific research and practical applications such as new drug development. A variety of techniques are now available to experimental biologists for discovering protein–protein interactions, such as yeast two-hybrid systems (Ito et al., 2000), mass spectrometry (Ho et al., 2002), and many others as surveyed in Shoemaker and Panchenko (2007a). Although these high-throughput experimental methods have accumulated a large amount of data, interactomes of many organisms are far from complete (Shoemaker and Panchenko, 2007a). The low interaction coverage, along with the experimental biases toward certain protein types and cellular localizations reported by most experimental techniques, call for the development of computational methods that are able to predict more reliable putative PPI for further experimental screening (Shoemaker and Panchenko, 2007b).
Computational methods for predicting protein interaction partners in early ages use genomic or protein context to infer functional associations (Shoemaker and Panchenko, 2007b). It is assumed in gene neighbor and gene cluster methods (Bowers et al., 2004b; Ermolaeva et al., 2001; Moreno-Hagelsieb and Collado-Vides, 2002; Strong et al., 2003; Salgado et al., 2000) that genes with closely related functions that encode potentially interacting proteins are often transcribed as a single unit—an operon. The phylogenetic profile methods (Bowers et al., 2004a, 2005; Pagel et al., 2004; Barker and Pagel, 2005) are based on the hypothesis that functionally linked and potentially interacting nonhomologous proteins coevolve and have orthologs in the same subset of fully sequenced organisms. The Rosetta Stone approaches (Marcotte et al., 1999; Enright et al., 1999; Marcotte and Marcotte, 2002; Yanai et al., 2001) infers protein interactions from protein sequences in different genomes. Sequence-based coevolution methods (Goh et al., 2000; Jothi et al., 2006; Ramani and Marcotte, 2003) take the perspective that interacting proteins very often coevolve so that changes in one protein leading to the loss of function or interaction should be compensated by the correlated changes in another protein. More in silico methods to predict protein–protein interactions are surveyed in Shoemaker and Panchenko (2007b).
Recently, machine-learning techniques, such as Bayesian networks (Jansen et al., 2003), decision trees (Zhang et al., 2004; Chen and Liu, 2005), random forest (Qi et al., 2005; Chen and Jeong, 2009), and support vector machines (SVM) with different kernels (Martin et al., 2005; Ben-Hur and Noble, 2005; Shen et al., 2007; Martial et al., 2010), have been successfully applied to predict PPIs. These methods used various data sources to train a classifier to distinguish positive examples of truly interacting protein pairs from the negative examples of noninteracting pairs. However, all these methods suffer from a fundamental difficulty—how to choose the negative samples. Compared to the obvious choice of positive samples from truly interacting protein pairs, negative samples are typically hard to be chosen. First, noninteracting protein pairs refer to those currently without experimental or computational evidence to support a physical interaction or functional association. In reality, however, such protein pairs could interact. Second, the number of noninteracting protein pairs is much larger than the number of the interacting ones, therefore, unbalanced training data often cause skewed prediction models that lead to unsatisfactory prediction results. In most existing classification-based methods, heuristic ways have been employed to tackle these problems.
With the above recognitions, instead of considering PPI prediction as a classification problem, we approach it from a different perspective using matrix completion, which is an important mathematical topic to address the problem to recover a matrix from what appears to be incomplete or even corrupted (Candès and Plan, 2009). An illustrative example to predict missing PPIs using matrix completion is given in Figure 1. Given a protein interaction network, a graph can be naturally constructed as shown in the top left panel of Figure 1, with vertices representing proteins and edges representing known PPIs. The adjacency matrix of the resulted graph is shown in the bottom left panel of Figure 1, where “1” indicates a known PPI and “?” indicates that there currently exists no experimental evidence to support a physical interaction between the corresponding protein pair. Our task is to identify putative interacting protein pairs from those marked as “?”. Using a matrix completion algorithm, the unknown entries of the matrix is filled as in the bottom right panel of Figure 1, where the value filled for an unknown entry indicates how likely it should be recovered. As a result, a list of ranking scores are produced for the noninteracting protein pairs, and prediction can be performed by picking up the top-ranking ones as putative PPIs. As shown in the top right panel of Figure 1, the top three ranked unknown entries are predicted as putative PPIs, which are marked as blue dash lines and correspond to the underlined entries in the filled matrix in the bottom right panel of Figure 1. Obviously, matrix completion only uses truly interacting protein pairs without requiring negative training samples, thus the difficulty in existing classification-based PPI prediction methods is circumvented.
Prediction of putative protein–protein interactions (PPIs) can be performed as a process of matrix completion. Top left: Original PPI graph. Bottom left: Adjacency matrix of the original PPI graph, where “1” indicates a known PPI and “?” indicates the protein pair has no experimental support to have a physical link. Bottom right: A process of matrix completion. Top right: Predict missing PPIs by taking the entries with top 3 ranking scores in the filled matrix.
Another important property of protein interaction networks makes matrix completion of particular use in predicting PPIs. Recent studies (Valdar and Thornton, 2001; Teichmann, 2002; Nooren and Thornton, 2003; Aloy et al., 2003; Littler and Hubbard, 2005; Panchenko et al., 2005; Dai and Prasad, 2010) have confirmed that, as opposed to the huge number of protein interactions, the number of interaction types or modes is limited and rather small, (i.e., the adjacency matrices of PPI graphs by nature are low-rank matrices). It is also proved in mathematics (Candès and Recht, 2009) that if the unknown matrix is known to have low rank or approximately low rank, accurate and even exact matrix recovery is possible. Therefore, using matrix completion methods, putative protein interactions can be inferred from incomplete, or even noisy, input PPI graphs.
In this article, we propose a novel nonnegative matrix tri-factorization (NMTF)-based (Lee and Seung, 1999; Ding et al., 2006b) matrix completion approach to predict candidate protein–protein interactions. NMTF focuses on the analysis of data matrices whose elements are nonnegative, such as the adjacency matrix of a PPI graph, and decomposes the input matrix into three nonnegative factor matrices that approximate the input matrix by a low-rank nonnegative representation (Ding et al., 2005, 2006a), which, due to its mathematical elegance, has been widely studied (Wang et al., 2011d) and applied to solve a variety of real-world problems (Wang et al., 2011b, 2011c). We first employ NMTF approach to predict putative protein interactions, which only makes use of the PPI network data. After that, we extend the standard NMTF framework by adding manifold regularization (Gu and Zhou, 2009), such that additional biological data, for example, protein sequences data, protein structures information, and gene expressions, can be incorporated to achieve enhanced PPI prediction performance. Our extension for manifold regularization is different from existing works (Cai et al., 2008; Gu and Zhou, 2009) where we emphasize the orthogonality on the factor matrices, which avoids degenerate solutions and makes our method more robust to parameter selection. Extensive empirical evaluations on four different genomic species have shown encouraging performance, which demonstrate the effectiveness of the proposed methods.
2. Methods
First we briefly formalize the problem of PPI prediction. Given a PPI network, we may construct a graph G = (V, Ω, X), with V corresponding to n = |V| proteins and Ω ⊆ V × V corresponding to known PPIs. \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${{\bf X}} \in \{0 , 1 \} ^{n \times n}$$
\end{document} is the adjacency matrix, such that Xij = 1 if \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$(i , j) \in \Omega$$
\end{document} (i.e., there exists a PPI between protein i and protein j), and Xij = 0 otherwise. Our task is to identify a subset of noninteracting protein pairs M ⊆ (V × V)\Ω, which tend to interact and can be served as potential targets for further experimental screening.
Throughout this article, we denote matrices as boldface, uppercase characters. Given a matrix M, its Frobenius norm and trace are denoted as ∥M∥ and tr (M) respectively. For convenience, given a index set M of a matrix X, we define XM as following:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}({\bf X}_M) _{ij} = \begin{cases}{\bf X}_{ij} , \quad \ \forall \ (i , j) \in M , \\ 0 \quad \qquad {\rm otherwise.}\end{cases} \tag{1}\end{align*}
\end{document}
2.1. Predict new protein interactions via PPI networks
Objective to predict PPIs We first predict protein interactions only using PPI network data. Considering the protein interaction prediction as a matrix completion problem, where the input PPI adjacency matrix X contains missing entries (pairs of proteins whose interactions are yet to be determined), we wish to predict Y, which has full entries (i.e., every element of Y is filled with computed values). Y completes X in the sense that YΩ = XΩ, or more explicitly, Yij = Xij, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\forall (i , j) \in \Omega$$
\end{document}, where Ω denotes the set of edges where the input adjacency matrix X has known values (the set of interacting edges). Mathematically, the PPI prediction problem can be solved as the following optimization problem:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}\min \limits_{\bf {Y}} \ J_1 = \ \mid\mid {\bf X} - {\bf Y} \mid\mid _ \Omega^2 = \sum \limits_{(i , j) \in \Omega} ({\bf X} - {\bf Y}) _{ij}^2. \tag{2}\end{align*}
\end{document}
Due to the low-rank nature of the adjacency matrix as discussed earlier, the completed matrix Y can be factorized and written as Y = HSHT, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\bf H} \in {\mathbb R}_{+} ^{n \times k}$$
\end{document} and \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\bf S} \in {\mathbb R}_{+} ^{k \times k}$$
\end{document} are the factor matrices with nonnegative elements. As a result, Y = HSHT can be seen as a low-rank representation of the input matrix X with rank of k ≪ n. Thus, we can rewrite Equation (2) as follows:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}\min \limits_{{\bf H} \geq 0 , {\bf S} \geq 0} \ J_2 = \mid\mid {\bf X} - {\bf HSH}^T \mid\mid _ \Omega^2. \tag{3}\end{align*}
\end{document}
Note that, although there exist other low-rank matrix approximation methods, for example, singular value decomposition (SVD), using NMTF as in Equation (3) to constrain the factor matrices H and S to be nonnegative is a natural choice because all the entries of the input PPI adjacency matrix X are positive by definition. Moreover, because of the clustering interpretation of NMTF (Ding et al., 2005, 2006b), other biological data sources can be easily incorporated via manifold regularization as introduced later.
The solution algorithm Different from standard NMTF-based objectives (as in Lee and Seung, 1999; Ding et al., 2005, 2006b; Wang et al., 2011a), which are defined over the whole input nonnegative matrix, the objective in Equation (3) for PPI prediction is defined over a subset of the entries that correspond to known PPIs. Therefore, the solution algorithms to standard NMTF cannot be directly applied to solve Equation (3). To this end, we present an iterative algorithm in Algorithm 1 to solve Equation (3).
The main step of Algorithm 1 is step 4, which solves a symmetric NMF problem (Wang et al., 2011a). Following our earlier publication in Wang et al. (2011a), Equation (5) can be solved by an iterative algorithm with the following updating rules:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*} {\bf H} _ {jk} \leftarrow {\bf H} _ {jk} {\left[\frac {{\bf (ZHS)} _ {jk}} {({\bf HSH} ^T {\bf HS}) _ {jk}} \right] ^ {1 / 4}} , \qquad {\bf S} _ {ik} \leftarrow {\bf S} _ {ik} \frac {({\bf H} ^T {\bf ZH}) _ {ik}} {({\bf H} ^T {\bf HSH} ^T {\bf H}) _ {ik}} , \tag {4} \end{align*}
\end{document}
where the superscripts are removed for notation brevity. The correctness and convergence of the updating rules in Equation (4) can be rigorously proved as detailed in Wang et al. (2011a). Apparently, Algorithm 1 converts the difficult problem to solve Equation (3) as a series of standard NMTF problems, for which existing algorithms, such as the updating rules in Equation (4), can be used.
Solving Equation (3) by Algorithm 1 for matrix completion, our NMTF approach to predict PPIs is proposed.
The convergence of the solution algorithm Now we prove the following theorem that guarantees the convergence of Algorithm 1.
As in Equation (10), the approximation errors, that is, the objective value J1 in Equation (2), goes down monotonically but remains bigger than zero, i.e., \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mid \mid {\bf X} - {\bf Y}^{(0)} \mid \mid ^2_ \Omega \geq \mid \mid {\bf X} - {\bf Y}^{(1)} \mid \mid ^2_ \Omega \geq \mid \mid {\bf X} - {\bf Y}^{(2)} \mid \mid ^2_ \Omega \geq \cdots \geq 0$$
\end{document}, therefore Theorem 1 guarantees the convergence of the algorithm.
2.2. Predict new protein interactions from multimodal biological data
In the last subsection, we introduced how to infer putative protein interactions using only the PPI network data, while in practice we may have other biological data as well, such as protein sequence data (Martin et al., 2005; Shen et al., 2007; Martial et al., 2010), 3D protein structures (Ben-Hur and Noble, 2005; Chen and Liu, 2005; Qiu et al., 2007), and so on. To exploit the useful information contained in the biological data from other sources, in this subsection we further develop the proposed NMTF-based matrix completion approach.
An important reason for the popularity of NMTF in statistical learning lies in its close connection to k-means clustering (Ding et al., 2005, 2006b). Specifically, given a symmetric nonnegative input matrix W, the factor matrix H can be seen as the clustering indications of the vertices (Luo et al., 2009). Therefore, if we have biological data other than PPI networks appearing in form of pairwise similarity, we can incorporate them through manifold regularization (Chung, 1997; Shi and Malik, 2000). Specifically, let \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\bf W}_{(k)} (0 \leq k \leq K)$$
\end{document} be a set of pairwise similarities constructed from different biological data, an integrated similarity among proteins can be constructed as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\bf W} = \sum \nolimits_k \eta_k{\bf W}_{(k)} \ {(\eta_k \geq 0, \sum \nolimits_k \eta_k = 1)}$$
\end{document} where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\eta_k$$
\end{document} are parameters to balance the data from different sources. We further develop the objective in Equation (3) as follows:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}\min \limits_{{\bf H} \geq 0 , {\bf S} \geq 0} J_4 = \mid \mid {\bf X} - {\bf HSH}^T \mid \mid _ \Omega^2 + 2 \lambda {\bf tr} ({\bf H}^T ({\bf D} - {\bf W}) {\bf H)} , \qquad s.t. \ {\bf H}^T{\bf D}{\bf H} = I , \tag{11}\end{align*}
\end{document}
where D is a diagonal matrix whose diagonal entries Dii = ∑jWij are the degree of the corresponding data points on W, and λ is a parameter to balance the relative importance of the regularization term, which is empirically selected as λ = 0.01 in all our experimental evaluations. Because H can be seen as the “soft” clustering labels (Ding et al., 2005), the second term in Equation (11) enforces the smoothness over the variation of the clustering labels with respect to the underlying manifold described by W (Cai et al., 2008; Gu and Zhou, 2009), by which additional biological data sources are incorporated.
Equation (11) takes a similar form to Equation (3), which, again, is not a standard NMTF problem. We use Algorithm 1 to solve it by replacing step 4 to minimize the following objective:
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
\begin{align*}J_4 = \mid \mid {\bf Z} - {\bf HSH}^T \mid \mid ^2 + 2 \lambda{\bf tr} ({\bf H}^T ({\bf D} - {\bf W}) {\bf H}) , \qquad s.t. \ {\bf H} \geq 0 , {\bf S} \geq 0 , {\bf H}^T{\bf DH} = I , \tag{12}\end{align*}
\end{document}
where Λ is the Lagrangian multiplier for the constraint HTDH = I, and its value is given by Λ =HTXHS − HTHSHTHS + λHTWH. The rigorous proof of the correctness and the convergence of the iteration procedures in Equation (13) can be found in our earlier publication in Wang et al. (2011e).
Note that, the objective in Equation (12) is different from the objectives of many existing related works (Cai et al., 2008; Gu and Zhou, 2009) using NMF in that the orthogonality HTDH = I is enforced. This is important because the second term of Equation (12) can be written as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\bf tr} ({\bf H}^T ({\bf D} - {\bf W}) {\bf H}) = \sum\nolimits_{k = 1}^K h_k^T ({\bf D} - {\bf W}) h_k$$
\end{document}. Without the orthogonality constraint, different columns become independent of each other, thereby reaching the same minimum with same solution, that is, \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$h_1^* = \cdots = h_k^*$$
\end{document}. When λ is not small, degenerate solution will be obtained and the approximation of Z by HSHT is degraded. We refer interested readers to Wang et al. (2011e) for more detailed discussions on the importance and effectiveness of enforcing orthogonality constraints in manifold regularized NMTF.
Solving Equation (11) for matrix completion, our regularized non-negative matrix tri-factorization (R-NMTF) approach for PPI prediction is proposed, which is able to utilize both PPI network data as well as other biological data.
3. Materials And Data Sources
Protein interaction networks We construct PPI graphs using the protein interaction networks compiled by BioGRID database (Stark et al., 2006). We evaluate our methods on the four species as follows: Saccharomyces cerevisiae, Drosophila melanogaster, Homo sapiens, and Caenorhabditis elegans. For each species, an undirected graph is constructed, with vertices representing proteins and edges representing observed physical interactions. For each graph, we only consider the largest connected component of the physical interaction map from BioGRID database of version 2.0.56. The details of the four PPI graphs are listed in Table 1, where “coverage” computes the percentage of known PPIs against the total number of protein pairs (n × (n − 1)/2).
PPI Graphs of Four Species Constructed Using BioGRID Database of Version 2.0.56
S. cerevisiae
D. melanogaster
H. sapiens
C. elegans
Proteins
5056
7294
8255
3353
Edges (number/coverage)
9439/0.738%
24960/0.094%
25929/0.076%
6449/0.114%
Protein sequence data We download protein sequence data from GenBank (Benson et al., 2006) and compute the sequence based similarity using the mismatch kernel (Leslie et al., 2003). A protein sequence si is first mapped to a feature vector \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\Phi_{k , m}{(s_i)} = \{\phi_ \beta{(\alpha)} \} _{\beta \in {\cal A}^k}$$
\end{document}, where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal A}$$
\end{document} is the alphabet of 20 amino acids. The neighborhood \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal N}_{k , m} (\alpha)$$
\end{document} of a k-mer α is the set of k-mers that differs in at most m positions. The feature vector encodes all the k-mers in the neighborhood for \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\phi_ \beta (\alpha) = 1 \ {\rm if} \ \alpha \in {\cal N}_{k , m} (\beta)$$
\end{document}, and 0 otherwise. Then the mismatch kernel, thereby the induced pairwise similarity between two protein sequences si and sj, is computed as \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$W_{ij}^{(1)} = {\cal K}{(s_i , s_j)} = \langle \Phi_{k , m}{(s_i)} , \Phi_{k , m}{(s_j)} \rangle$$
\end{document}. In our empirical studies, we set k = 6 and m = 1, which is the same as in Martial et al. (2010). In our empirical studies, protein sequence data are used as the addition biological data source, that is, W = W(1).
Protein annotation data We use the functional annotations from Gene Ontology (GO) Consortium (Ashburner et al., 2000), which is a set of structured vocabularies organized in a rooted directed acyclic graph (DAG), describing attributes of gene products (proteins or RNA) in three categories of “cellular component,” “molecular function,” and “biological process.”
4. Experimental Results And Discussions
4.1. Improved prediction capability in cross-validation
We first evaluate the proposed methods and compare their prediction capabilities against three most recent PPI prediction methods.
(1) Tensor product pairwise kernel (TPPK) method (Ben-Hur and Noble, 2005): This method builds a kernel for pairwise objects. In order for a fair comparison, protein sequence and protein interaction network topology are used for kernel construction. PPI prediction is then carried out by the ranking scores for noninteracting protein pairs yielded by an SVM on the resulted score.
(2) Metric learning pairwise kernel (MLPK) method (Qiu et al., 2007): This method represents a pair of objects as the difference between its members, such that the resulted kernel is invariant with respect to the order of the proteins. Again, SVM is used to compute the ranking score for putative PPIs.
(3) Nearest neighbor (NN) (Martial et al., 2010) method: NN is the simplest classification method in machine learning. In Martial et al. (2010), a ranking score for each noninteracting protein pair is computed as
where \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$${\cal N}_k (x_i)$$
\end{document} is the set of k-nearest neighbors of xi, and d (·,·) is distance function built from a kernel by \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$d (x_i , x_j) = \sqrt{{\cal K} (x_i , x_i) - 2{\cal K} (x_i , x_j) + {\cal K} (x_j , x_j)}$$
\end{document}. In our evaluations, we use the mismatch kernel for protein sequence data.
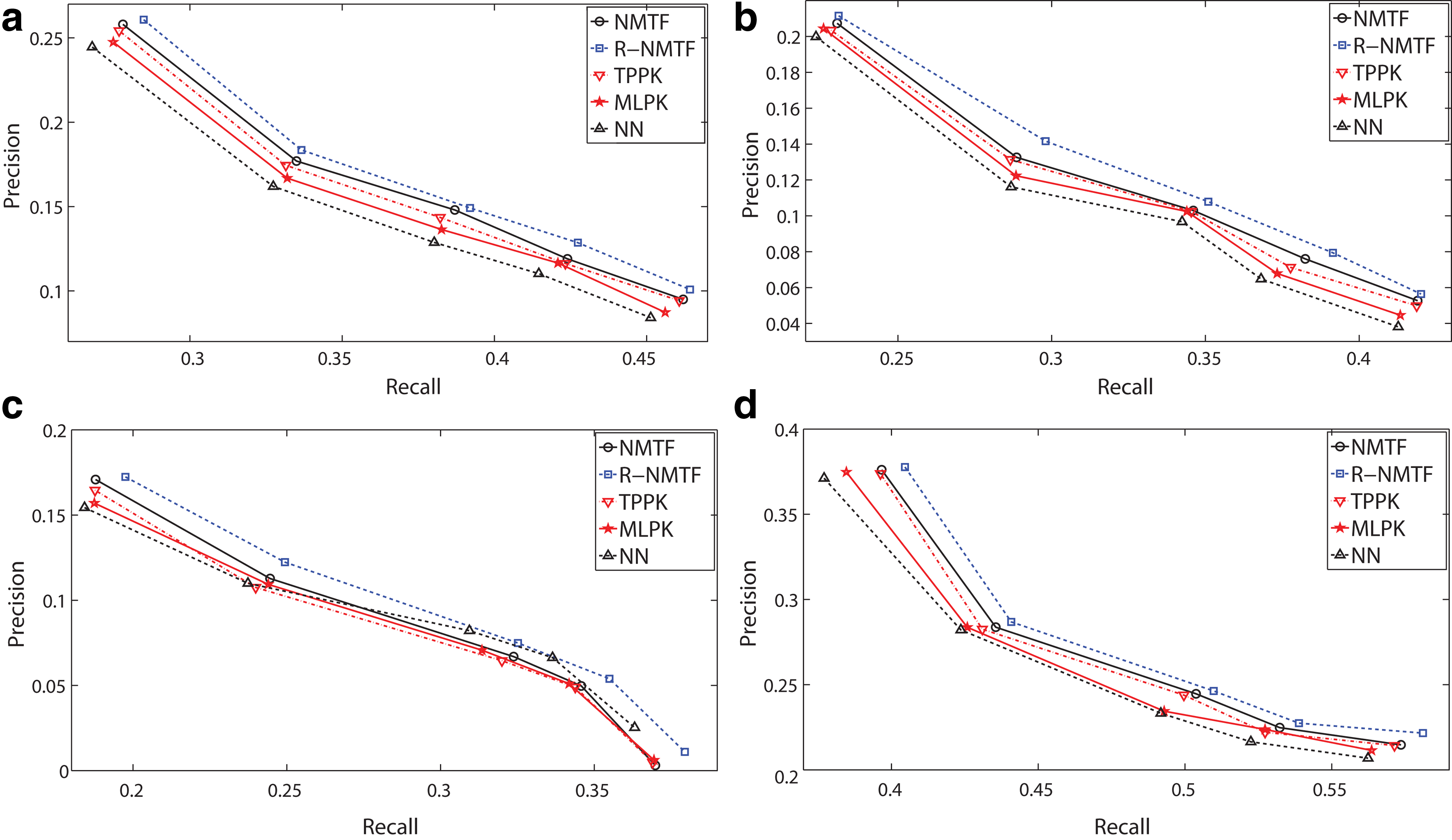
Experimental procedures For each method, we perform 20-fold cross-validation as follows. For each trial, we remove 5% known edges (PPIs) from the input graph and try to recover them using the remaining graph, which is repeated by 10 times. The average results over the 10 trials on every species are reported in Figure 2. During each trial, an internal five-fold cross validation is performed for parameter selection. For our NMTF and R-NMTF methods, the parameter is the rank k of the factor matrices H and S. For TPPK and MLPK methods, we use the Gaussian kernel, therefore the parameters are the two regularization parameters. For NN method, we select the k of NN from {1, 2, 3, 5, 10, 15}, which is the same as in Martial et al. (2010). We fine-tune the parameters for best prediction precision for all the compared methods.
Precision-recall curves by 20-fold cross-validation on four species by the compared PPI prediction methods. NMFT, nonnegative matrix tri-factorization; R-NMTF, regularized nonnegative matrix tri-factorization; TPPK, tensor product pairwise kernel method; MLPK, metric learning pairwise kernel method; NN, nearest neighbor.
Results Because all compared methods produce a list of ranking scores for noninteracting protein pairs, we employ precision-recall curves to measure the prediction performance. We compute the precisions and recalls when picking up a range of top k noninteracting protein pairs as predictions and average them over the 10 trials. The resulted precision-recall curves of four species are reported in Figure 2. From the results, we can see that both proposed methods, NMTF and R-NMTF, consistently outperform comparable methods, sometimes very significantly. In addition, the prediction performances of R-NMTF method are always better than those of NMTF method, which is consistent with our previous theoretical analysis in that multimodal biological data, i.e., protein interaction network plus protein sequence data, offer enhanced prediction performance. We also observe that the TPPK method achieves similar performances to the proposed NMTF method for D. melanogaster and C. elegans species. However, our NMTF method uses only protein interaction network data, while TPPK method exploits both network data and sequence data.
A more careful analysis on the prediction results shows that the noninteracting protein pairs (including the noninteracting protein paris in the original PPI graphs and those removed due to cross-validation) with high ranking scores identified by the proposed methods typically exhibit high similarities in their functional roles. In Table 2, we list the predicted protein pairs with top 10 highest ranking scores by R-NMTF method on S. cerevisiae species, in which the biological functions of all protein pairs are very similar to each other. For example, “PHO91” works as “Low-affinity phosphate transporter of the vacuolar membrane,” which is also the main functional role of its putative interacting partner “PHO90.” Moreover, both of them have functionalities of “transcription independent of Pi and Pho4p activity” and “overexpression results in vigorous growth.” These observations clearly demonstrate that these two proteins are functionally related and tend to interact with each other, which provides concrete evidence to support that the predicted protein interactions by the proposed R-NMTF are biologically meaningful.
Comparisons of Biological Functionalities of the Predicted Protein Interactions
LAT1
PDX1
Dihydrolipoamide acetyltransferase component (E2) of pyruvate dehydrogenase complex, which catalyzes the oxidative decarboxylation of pyruvate to acetyl-CoA—Lat1p; protein coding
Dihydrolipoamide dehydrogenase (E3)-binding protein (E3BP) of the mitochondrial pyruvate dehydrogenase (PDH) complex, plays a structural role in the complex by binding and positioning E3 to the dihydrolipoamide acetyltransferase (E2) core—Pdx1p; protein coding
PHO91
PHO90
Low-affinity phosphate transporter of the vacuolar membrane; deletion of pho84, pho87, pho89, pho90, and pho91 causes synthetic lethality; transcription independent of Pi and Pho4p activity; overexpression results in vigorous growth—Pho91p; protein coding
Low-affinity phosphate transporter; deletion of pho84, pho87, pho89, pho90, and pho91 causes synthetic lethality; transcription independent of Pi and Pho4p activity; overexpression results in vigorous growth—Pho90p; protein coding
PHO91
PHO87
Low-affinity phosphate transporter of the vacuolar membrane; deletion of pho84, pho87, pho89, pho90, and pho91 causes synthetic lethality; transcription independent of Pi and Pho4p activity; overexpression results in vigorous growth—Pho91p; protein coding
Low-affinity inorganic phosphate (Pi) transporter, involved in activation of PHO pathway; expression is independent of Pi concentration and Pho4p activity; contains 12 membrane-spanning segments—Pho87p; protein coding
PHO87
PHO89
Low-affinity inorganic phosphate (Pi) transporter, involved in activation of PHO pathway; expression is independent of Pi concentration and Pho4p activity; contains 12 membrane-spanning segments—Pho87p; protein coding
Na+/Pi cotransporter, active in early growth phase; similar to phosphate transporters of Neurospora crassa; transcription regulated by inorganic phosphate concentrations and Pho4p—Pho89p; protein coding
COY1
SVP26
Coy1p—Golgi membrane protein with similarity to mammalian CASP; genetic interactions with GOS1 (encoding a Golgi snare protein) suggest a role in Golgi function; protein coding
Integral membrane protein of the early Golgi apparatus and endoplasmic reticulum, involved in COP II vesicle transport; may also function to promote retention of proteins in the early Golgi compartment—Svp26p; protein coding
HOM3
HOM2
Aspartate kinase (L-aspartate 4-P-transferase); cytoplasmic enzyme that catalyzes the first step in the common pathway for methionine and threonine biosynthesis; expression regulated by Gcn4p and the general control of amino acid synthesis—Hom3p; protein coding
Aspartic beta semi-aldehyde dehydrogenase, catalyzes the second step in the common pathway for methionine and threonine biosynthesis; expression regulated by Gcn4p and the general control of amino acid synthesis—Hom2p; protein coding
REC114
REC102
Protein involved in early stages of meiotic recombination; possibly involved in the coordination of recombination and meiotic division; mutations lead to premature initiation of the first meiotic division—Rec114p; protein coding
Protein involved in early stages of meiotic recombination; required for chromosome synapsis; forms a complex with Rec104p and Spo11p necessary during the initiation of recombination—Rec102p; protein coding
PCF11
REF2
Pcf11p—mRNA 3’ end processing factor, essential component of cleavage and polyadenylation factor IA (CF IA), involved in pre-mRNA 3’ end processing and in transcription termination; binds C-terminal domain of largest subunit of RNA pol II (Rpo21p); protein coding
RNA-binding protein involved in the cleavage step of mRNA 3’-end formation prior to polyadenylation, and in snoRNA maturation; part of holo-CPF subcomplex APT, which associates with 3’-ends of snoRNA- and mRNA-encoding genes—Ref2p; protein coding
DIM1
IMP4
Dim1p—Essential 18S rRNA dimethylase (dimethyladenosine transferase), responsible for conserved m6(2)Am6(2)A dimethylation in 3’-terminal loop of 18S rRNA, part of 90S and 40S pre-particles in nucleolus, involved in pre-ribosomal RNA processing; protein coding
Component of the SSU processome, which is required for pre-18S rRNA processing; interacts with Mpp10p; member of a superfamily of proteins that contain a sigma(70)-like motif and associate with RNAs—Imp4p; protein coding The BioGRID Database Seperator
FCY22
FCY2
Fcy22p—Putative purine-cytosine permease, very similar to Fcy2p but cannot substitute for its function; protein coding
Fcy2p—Purine-cytosine permease, mediates purine (adenine, guanine, and hypoxanthine) and cytosine accumulation; protein coding The BioGRID Database Seperator
Note that, in our empirical studies, we only use one additional data source (i.e., protein sequence data) for the purpose of demonstration. In practice, more biological data, when available, could be incorporated through a proper kernel construction under the R-NMTF prediction framework to achieve better prediction results.
4.2. Conserved functional similarity of predicted new protein interactions
It is generally believed that interacting protein pairs tend to have similar functional roles. We test this hypothesis by evaluating the function similarities between our predicted interacting proteins on S. cerevisiae species. We use the Resnik score (Resnik, 1995) on the GO annotation system to measure the functional similarities between protein pairs.
Specifically, given a term t in GO, its information content is quantified as −log, where the probability of the term is taken to be its frequency among annotations to all GO terms. If a term t is annotated to a protein, it must be annotated with all the ancestors of the term t. Therefore, the higher the term is located in the GO hierarchy, the less its information content. In the case of GO, a term might have multiple parent terms, so that a pair of terms might have more than one path of common ancestors. Denoting the set of all common-ancestor terms of t1 and t2 as A(t1, t2) we define the similarity between two terms, t1 and t2, as sim \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$ (t_1, t_2) = \max \nolimits_{a \in A} (t_1, t_2) - \log p(a) $$
\end{document}. Because an individual protein could have multiple functions, we compute the similarity of two proteins, p1 and p2, by matching each function of p1 to its most similar function of p2, and the average is taken over all such pairs of functions as sim \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$(p_1 \rightarrow p_2) = {\rm avg} \left[\sum{_{s \in p_1}} \max \nolimits_{{t \in p_2}} \ {\rm sim} (s , t) \right]$$
\end{document}. As this measurement is not symmetric with respect to p1 and p2, the final semantic similarity between the two proteins is defined as sim \documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$(p_1 , p_2) = \frac {1} {2} \times \left[{\rm sim} \ (p_1 \rightarrow p_2) + {\rm sim} \ (p_2 \rightarrow p_1) \right]$$
\end{document}.
The average Resnik scores of known interacting protein pairs, noninteracting protein pairs, and predicted putative interacting protein pairs by the proposed NMTF and R-NMTF methods are reported in Table 3, from which we have the following observations.
Resnik Scores of Different Protein Pairs
Average ± standard deviation
Known interacting protein pairs
7.160 ± 1.321
Noninteracting protein pairs
3.481 ± 1.129
Predicted putative interacting protein pairs by NMTF method
6.246 ± 1.031
Predicted putative interacting protein pairs by R-NMTF method
First, the average Resnik score of known interacting protein pairs is higher than that of noninteracting protein pairs, which is consistent with the aforementioned assumption that the interacting protein pairs have more similar biological functions than those not interacting.
Second, the average Resnik score of the predicted putative interacting protein pairs by the proposed NMTF and R-NMTF methods are close to that of known interacting protein pairs. Namely, the proteins in the predicted PPIs are highly functionally similar, which confirms the correctness of the proposed methods from protein function similarity perspective.
Last, but not least, the predicted protein interactions by R-NMTF method have higher semantic score than those by NMTF method. Therefore, prediction by R-NMTF method using multimodal biological data sources is more advantageous than that by NMTF method using only one data source.
4.3. Capability to predict new protein interactions
The ultimate goal of computational methods to predict protein interactions is to discover new interacting protein pairs that can be served as potential targets for experimental screening. Therefore, we predict protein interactions on S. cerevisiae species by the proposed R-NMTF method on the PPI graph using BioGRID data of version 2.0.56 (published on August 31, 2009).
We examine the prediction results and find that among the top 200 putative PPIs predicted by our R-NMTF method, 87 protein pairs are consistent with other evidence (i.e., they are verified by experimental results and appear in recent published literatures). For example, protein “BNI1” and protein “CTF3” are not documented as a PPI in BioGRID data of version 2.0.56 but predicted to be interacting by our method. By a careful document survey, we notice that the experimental supporting document to this protein pair only appears in early 2010 (Vizeacoumar et al., 2010). This result firmly confirms the effectiveness of our method in predicting new protein interactions. Other predicted putative protein pairs together with the PubMed document IDs for their supporting literatures are listed in AppendixTable 5. Most them are also incorporated in the most recent BioGRID data of version 3.1.69 (published on September 30, 2010). The high overlaps between our predictions and the results in existing literature give a solid support of the usefulness of the proposed method.
4.4. Improved protein function prediction using predicted PPI networks
Protein interaction networks are broadly used in various biological applications, whose performances are inevitably determined by the quality input PPI graphs. Therefore, we assess the quality of predicted PPI networks in protein function prediction on S. cerevisiae species.
We predict protein functions on the original PPI graph constructed from the BioGRID database, the PPI graph filled by the top 1000 putative interacting protein pairs predicted by NMTF method, and that by R-NMTF method. We make predictions using the following three benchmark graph-based protein function prediction methods:
(1) Majority voting (MV) (Schwikowski et al., 2000) method: This method assigns functions to a protein via its connecting neighbors in certain ranges.
(2) Iterative majority voting (IMV) (Vazquez et al., 2003) method: This method is the same as the MV method, but iteratively repeats the function assignment process until certain conditions are satisfied.
(3) Function flow (FF) (Nabieva et al., 2005) method: This method formulates the annotation problem as a minimum multiway-cut problem, where the goal is to assign a unique function to all unannotated proteins so as to minimize the cost of edges connecting proteins with different assignments.
We implement these methods following the details in the original literatures. Because FF method produces a ranking list of predicted protein functions, we select a threshold such that the prediction precision is maximized. Five-fold cross-validation is performed to predict the functions in “biological process” of GO. The average prediction precision over all test functions and five trials of cross-validation of the involved methods on different PPI graphs are reported in Table 4.
Performance of Protein Function Prediction by Involved Method on Compared PPI Graphs
The results in Table 4 show that the function prediction performance for all three methods are improved when the predicted PPI graphs are used. Such results experimentally prove that the predicted PPI graphs have higher quality than the original one, which demonstrates that the filled putative protein interactions by the proposed methods are largely biologically meaningful. Thus, we can tentatively conclude that the proposed NMTF and R-NMTF indeed can improve the protein interaction networks. Again, multimodal biological data sources based R-NMTF method is better than single data source-based NMTF method.
5. Conclusions
In this article, instead of considering protein–protein interaction as a binary classification problem, as in many existing works, we formulated it as a matrix completion problem. Taking this different perspective, the difficulty of selecting negative training samples in classification-based methods is averted. Moreover, because the number of protein interaction types is small, recovery of missing PPIs from an incomplete observed protein interaction network can be suitably solved under the framework of matrix completion. We first proposed to use NMF approach to predict PPIs from protein interaction network data, and then extended it through manifold regularization to incorporate multimodal biological data sources. We have conducted extensive empirical studies to evaluate different aspects of the proposed methods on four genomic species including S. cerevisiae, D. melanogaster, H. sapiens, and C. elegans. Promising results in the experiments validate our methods, which are consistent with our theoretical analysis.
6. Appendix
6.1. Predicted new protein interactions with highest-ranking scores by R-NMTF method
In Table 5, we list a subset (87 out of 200) of the predicted putative protein interactions with highest ranking scores on the original S. cerevisiae PPI graph by the proposed R-NMTF method. These protein pairs are already discovered to be interacting in existing literatures. The document ID in PubMed of the supporting literatures are listed in the third column of Table 5.
Predicted PPIs with Highest Ranking Scores by R-NMTF Method
Interacting protein A
Interacting protein B
PubMed ID of the supporting literature
BNI1
CTF3
20065090
ACS2
RTC1
18676811
BNA4
VHS3
20093466
ALG3
ERG2
19325107
APQ12
NUR1
20093466
AVT5
ELP2
20093466
BOS1
YDR186C
20093466
BMS1
IPT1
20093466
ATP5
PTC1
20093466
ATP5
CHK1
20093466
ARF1
GET2
16269340
AFT1
RPN4
20439772
BCY1
NFT1
20093466
ANP1
VID22
20093466
ANP1
MKK1
20093466
BEM1
UBP14
20093466
BNI1
MXR2
20065090
ATG8
APE2
20093466
ARP8
BUD13
20041197
ARP8
ESC2
20041197
ARO7
CSF1
20093466
ARP8
GSG1
20041197
ALG8
SPF1
19325107
ALP1
ARG1
16941010
ATP14
MDM35
20093466
AFG3
PET8
20093466
BIM1
RAD55
20065090
ARP9
PYC2
20093466
BAT1
SGF29
20093466
BRE2
ESBP6
20093466
ANP1
HXK2
20093466
APC5
RPN10
20093466
ABD1
MET6
20093466
ACT1
RIM101
20093466
ARO2
DTD1
20093466
BRE5
RPS10A
20508643
BUD14
PIG1
19841731
ABD1
PAC1
20093466
BET5
GSG1
19416478
BNI1
YPL066W
20093466
ARF1
IST2
20093466
BRE2
NUP188
20093466
ACH1
CIT1
20093466
AMD1
YJL070C
18719252
ARC18
PTC6
20093466
ATP18
ATP18
16716082
ARL1
MRN1
20093466
BNI4
VAC14
20093466
ARP1
KEM1
20093466
ALG8
APM3
20093466
BIM1
SAC6
20065090
ATP18
AEP2
20093466
BNI1
RTT109
20065090
ARP9
YJR079W
20093466
ALG9
SAP155
20093466
AHA1
SPT3
20093466
BCK1
MTC5
20093466
AAD3
MDM12
20093466
ALG9
SPT4
19325107
AIM22
IMP2
20093466
BRE5
TIF1
20508643
AVO1
GAA1
20101242
ARP8
URE2
20041197
BAT1
BCH1
20093466
BRR2
RAX1
20093466
AIM26
BCK1
14764870
APC5
MUM2
20093466
ALD5
OAR1
20093466
ALK2
LSM12
20489023
BNI1
DIP5
20065090
ALT1
MCX1
20093466
ACS2
OLA1
16429126
ALB1
TIF6
16651379
ATG21
INP53
20093466
BTS1
TLG2
19325107
APL1
SPG1
20093466
BRE5
HTB1
20508643
BIM1
RPL19B
20093466
ALG1
OPI3
20093466
ALB1
REI1
16651379
APS1
HOM6
20093466
AEP2
FYV10
20093466
AIM44
RAD5
20093466
BIM1
PET8
20065090
ARL3
ARL1
20093466
ANP1
PUS2
20093466
BOS1
YGR021W
20093466
References
1.
AloyP., CeulemansH., StarkA.et al.2003. The relationship between sequence and interaction divergence in proteins. J. Mol. Biol., 332:989–998.
2.
AshburnerM., BallC., BlakeJ.et al.2000. Gene ontology: tool for the unification of biology. Nat. Genet., 25:25–29.
3.
BarkerD., PagelM.2005. Predicting functional gene-links from phylogenetic-statistical analyses of whole genomes. PLoS Comp. Biol., 1:24–31.
4.
Ben-HurA., NobleW.2005. Kernel methods for predicting protein–protein interactions. Bioinformatics, 21:i38.
BowersP., CokusS., EisenbergD.et al.2004a. Use of logic relationships to decipher protein network organization. Science, 306:2246.
7.
BowersP., O'ConnorB., CokusS.et al.2005. Utilizing logical relationships in genomic data to decipher cellular processes. FEBS Journal, 272:5110–5118.
8.
BowersP., PellegriniM., ThompsonM.et al.2004b. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol., 5:R35.
9.
CaiD., HeX., WuX.et al.2008. Non-negative matrix factorization on manifold. In Proceedings of the 2008 Eighth IEEE International Conference on Data Mining, 63–72.
10.
CandèsE., PlanY.2009. Matrix completion with noise. Proceedings of the IEEE.
DaiO., PrasadN.2010. Low-rank matrix completion for inference of protein-protein interaction networks. In AIP Conference Proceedings. 1281:1531.
16.
DingC., HeX., SimonH.2005. On the equivalence of nonnegative matrix factorization and spectral clustering. Proc. SIAM Data Mining Conf, 606–610.
17.
DingC., LiT., JordanM.2006a. Convex and semi-nonnegative matrix factorizations for clustering and low-dimension representation. Technical Report LBNL-60428. Lawrence Berkeley National Laboratory.
18.
DingC., LiT., PengW.et al.2006b. Orthogonal nonnegative matrix t-factorizations for clustering. Proceedings of the 12th ACM SIGKDD international conference on knowledge discovery and data mining, 126–135.
19.
EnrightA., IliopoulosI., KyrpidesN.et al.1999. Protein interaction maps for complete genomes based on gene fusion events. Nature, 402:86–90.
20.
ErmolaevaM., WhiteO., SalzbergS.2001. Prediction of operons in microbial genomes. Nucleic Acids Res., 29:1216.
21.
GohC., BoganA., JoachimiakM.et al.2000. Co-evolution of proteins with their interaction partners1. J. Mol. Biol., 299:283–293.
22.
GuQ., ZhouJ.2009. Co-clustering on manifolds. Proceedings of the 15th ACM SIGKDD international conference on knowledge discovery and data mining, 359–368.
23.
HoY., GruhlerA., HeilbutA.et al.2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature, 415:180–183.
24.
ItoT., TashiroK., MutaS.et al.2000. Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proceedings of the National Academy of Sciences of the United States of America, 97:1143.
25.
JansenR., YuH., GreenbaumD.et al.2003. A Bayesian networks approach for predicting protein–protein interactions from genomic data. Science, 302:449.
26.
JothiR., CherukuriP., TasneemA.et al.2006. Co-evolutionary analysis of domains in interacting proteins reveals insights into domain-domain interactions mediating protein–protein interactions. J. Mol. Biol., 362:861–875.
27.
LeeD., SeungH.1999. Learning the parts of objects by non-negative matrix factorization. Nature, 401:788–791.
28.
LeslieC., EskinE., WestonJ.et al.2003. Mismatch string kernels for SVM protein classification. Adv Neural Inf Process Syst., 1441–1448.
29.
LittlerS., HubbardS.2005. Conservation of orientation and sequence in protein domain-domain interactions. J. Mol. Biol., 345:1265–1279.
30.
LuoD., DingC., HuangH.et al.2009. Non-negative laplacian embedding. In 2009 Ninth IEEE International Conference on Data Mining, 337–346.
31.
MarcotteC., MarcotteE.2002. Predicting functional linkages from gene fusions with confidence. Applied Bioinformatics, 1:93–100.
32.
MarcotteE., PellegriniM., NgH.et al.1999. Detecting protein function and protein–protein interactions from genome sequences. Science, 285:751.
33.
MartialH., MichaelR., Jean-PhilippeV.et al.2010. Large-scale prediction of protein–protein interactions from structures. BMC Bioinformatics, 11.
Moreno-HagelsiebG., Collado-VidesJ.2002. A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics, 18:S329.
36.
NabievaE., JimK., AgarwalA.et al.2005. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics, 21:i302.
37.
NoorenI., ThorntonJ.2003. Structural characterisation and functional significance of transient protein–protein interactions. J. Mol. Biol., 325:991–1018.
38.
PagelP., WongP., FrishmanD.2004. A domain interaction map based on phylogenetic profiling. J. Mol. Biol., 344:1331–1346.
39.
PanchenkoA., WolfY., PanchenkoL.et al.2005. Evolutionary plasticity of protein families: coupling between sequence and structure variation. Proteins: Structure, Function, and Bioinformatics, 61:535–544.
40.
QiY., Klein-SeetharamanJ., Bar-JosephZ.2005. Random forest similarity for protein–protein interaction prediction from multiple sources. Pac Symp Biocomput., 10:531–542.
41.
QiuJ., HueM., Ben-HurA.et al.2007. A structural alignment kernel for protein structures. Bioinformatics, 23:1090.
42.
RamaniA., MarcotteE.2003. Exploiting the co-evolution of interacting proteins to discover interaction specificity. J. Mol. Biol., 327:273–284.
43.
ResnikP.1995. Using information content to evaluate semantic similarity in a taxonomy. Arxiv preprint cmp-lg/9511007.
44.
SalgadoH., Moreno-HagelsiebG., SmithT.et al.2000. Operons in Escherichia coli: genomic analyses and predictions. Proceedings of the National Academy of Sciences of the United States of America, 97:6652.
45.
SchwikowskiB., UetzP., FieldsS.2000. A network of protein–protein interactions in yeast. Nat. Biotech., 18:1257–1261.
46.
ShenJ., ZhangJ., LuoX.et al.2007. Predicting protein–protein interactions based only on sequences information. Proceedings of the National Academy of Sciences, 104:4337.
47.
ShiJ., MalikJ.2000. Normalized cuts and image segmentation. IEEE. Trans. on Pattern Analysis and Machine Intelligence, 22:888–905.
48.
ShoemakerB., PanchenkoA.2007a. Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comp. Biol., 3:337–334.
49.
ShoemakerB., PanchenkoA.2007b. Deciphering protein–protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comp. Biol., 3:595–601.
50.
StarkC., BreitkreutzB., RegulyT.et al.2006. BioGRID: a general repository for interaction datasets. Nucleic Acids Res., 34:D535.
51.
StrongM., MallickP., PellegriniM.et al.2003. Inference of protein function and protein linkages in Mycobacterium tuberculosis based on prokaryotic genome organization: a combined computational approach. Genome Biol., 4:R59.
52.
TeichmannS.2002. The constraints protein-protein interactions place on sequence divergence. J. Mol. Biol., 324:399–407.
53.
ValdarW., ThorntonJ.2001. Protein-protein interfaces: analysis of amino acid conservation in homodimers. Proteins: Structure, Function, and Bioinformatics, 42:108–124.
54.
VazquezA., FlamminiA., MaritanA.et al.2003. Global protein function prediction from protein-protein interaction networks. Nat. biotechnology, 21:697–700.
55.
VizeacoumarF., Van DykN., VizeacoumarF.et al.2010. Integrating high-throughput genetic interaction mapping and high-content screening to explore yeast spindle morphogenesis. J. Cell. Biol., 188:69.
56.
WangH., HuangH., DingH.2011a. Simultaneous clustering of multi-type relational data via symmetric nonnegative matrix tri-factorization. In The 20th ACM Conference on Information and Knowledge Management.
57.
WangH., HuangH., NieF.et al.2011b. Cross-language web page classification via dual knowledge transfer using nonnegative matrix tri-factorization. In Proceedings of the 34th international ACM SIGIR conference (ACM SIGIR 2011), 933–942.
58.
WangH., NieF., HuangH., DingC.2011c. Dyadic transfer learning for cross-domain image classification. In 2011 IEEE International Conference on Computer Vision (IEEE ICCV 2011), 551–556.
59.
WangH., NieF., HuangH.et al.2011d. Fast nonnegative matrix tri-factorization for large-scale data co-clustering. In Twenty-Second International Joint Conference on Artificial Intelligence (IJCAI 2011).
60.
WangH., NieF., HuangH.et al.2011e. Nonnegative matrix tri-factorization based high-order co-clustering and its fast implementation. In 2011 Eleventh IEEE International Conference on Data Mining.
61.
YanaiI., DertiA., DeLisiC.2001. Genes linked by fusion events are generally of the same functional category: a systematic analysis of 30 microbial genomes. Proceedings of the National Academy of Sciences of the United States of America, 98:7940.
62.
ZhangL., WongS., KingO.et al.2004. Predicting co-complexed protein pairs using genomic and proteomic data integration. BMC Bioinformatics, 5:38.