
Abstract
Abstract
Recently a folding algorithm of topological RNA pseudoknot structures was presented in Reidys et al. (2011). This algorithm folds single-stranded γ-structures, that is, RNA structures composed by distinct motifs of bounded topological genus. In this article, we set the theoretical foundations for the folding of the two backbone analogues of γ structures: the RNA γ-interaction structures. These are RNA–RNA interaction structures that are constructed by a finite number of building blocks over two backbones having genus at most γ. Combinatorial properties of γ-interaction structures are of practical interest since they have direct implications for the folding of topological interaction structures. We compute the generating function of γ-interaction structures and show that it is algebraic, which implies that the numbers of interaction structures can be computed recursively. We obtain simple asymptotic formulas for 0- and 1-interaction structures. The simplest class of interaction structures are the 0-interaction structures, which represent the two backbone analogues of secondary structures.
1. Introduction
Interactions between nucleic acids can be understood to a good approximation at the level of secondary structures (patterns of base pairs) or extended secondary structures. From a technical point of view, therefore, the analysis, prediction, and comparison of RNA–RNA and RNA–DNA interaction structures are formalized as a suite of related combinatorial (optimization) problems that make genome-wide studies computationally feasible and set RNA bioinformatics apart from other areas of structural biology in which three-dimensional shapes and atomic detail cannot be neglected as easily. For instance, the interaction of eukaryotic miRNA with their targets, in addition to bacterial sRNA and mRNA—plant miRNA/mRNA and animal miRNA/mRNA interactions—may require a detailed 3D analysis by means of non-canonical base pairs. Despite this favorable starting point and the success of the combinatorial approach to structures of individual nucleic acid molecules, we nevertheless lack both theoretical insights and practical predictive power in the realm of interactions.
The simplest approach for folding RNA–RNA interaction structures concatenates two (or more) interacting sequences, one after another, remembering the specific merge point (cut-point), and then employs the standard secondary structure–folding algorithm on a single strand. This approach falls short of predicting many genuine features, such as kissing-hairpin loops. The paradigm of concatenation has also been generalized to include cross-serial interactions (Rivas and Eddy, 1999). The resulting model, however, still does not generate all relevant interaction structures (Busch et al., 2008). An alternative is to neglect any internal base pairings in either strand, that is, to compute the minimum free energy (MFE) secondary structure of hybridization of otherwise unstructured RNAs. This ansatz (Busch et al., 2008; Mückstein et al., 2006, 2008) restricts interactions to a single interval that remains unpaired in the secondary structure for each partner.
A different approach was taken independently by Alkan et al. (2006) and Pervouchine (2004), who proposed MFE folding algorithms for predicting the AP-structure of two interacting RNA molecules. In this model, the intramolecular structures of each partner are pseudoknot-free, the intermolecular binding pairs are noncrossing, and there is an ad hoc exclusion of so-called “zig-zag” motifs. The RNA–RNA interaction structures of Alkan et al. (2006), Bernhart et al. (2006), Hofacker et al. (1994), and Huang et al. (2010) have the following features: (a) When drawing the two backbones on top of each other, all base pairs are noncrossing, that is, no pseudoknots formed by internal or external arcs are allowed, and (b) zig-zag motifs are disallowed.
The abundance of RNA–RNA interactions is motivation to identify the analogue of RNA secondary structures. As in the case of RNA structures over one backbone, the key issue is to identify and analyze a suited filtration of RNA–RNA interaction structures. The filtration presented here is based on the topological genus of certain structural motifs and allows for a polynomial time folding of the accordingly filtered interaction structures. In contrast to a pure genus filtration, the structures presented here have arbitrarily high genus; it is only their motifs that are genus constrainted. The interaction structures considered here allow, in fact, for arbitrary composition and nesting of these genus-constrained motifs.
The classification and expansion of pseudoknotted RNA structures in terms of the topological genus of an associated fatgraph or double line graph were first proposed in Bon et al. (2008) and Orland and Zee (2002), although fatgraphs were applied to RNA secondary structures already by Penner (2004) and Penner and Waterman (1993). The first enumerative results initiated in Orland and Zee (2002) and Vernizzi and Orland (2005) are an application of matrix models from theoretical physics. Genus as well as other topological invariants of fatgraphs were introduced and studied as descriptors of proteins in Penner et al. (2010).
In Reidys et al. (2011), RNA γ-structures were introduced. The idea of γ-structures is to consider RNA structures obtained by building blocks, having topological genus at most γ. From this point of view, RNA secondary structures can be seen to be obtained by the building block that is simply an arc by means of concatenation and nesting. Accordingly, secondary structures are equivalent to 0 structures over one backbone. We call these blocks, in the case of genus, greater or equal to one irreducible shadow. In this sense, topological genus controls the complexity of the cross-serial interactions expressed in these irreducible shadows. In Reidys et al. (2011), but already implicit in Zagier (1995), it was proven that for any genus, there are only finitely many irreducible shadows.
RNA interaction structures of fixed topological genus (Andersen et al., 2012) have, as their one backbone counterpart, only finitely many irreducible shadows. Following the identification of these, Andersen et al. (2012) develops the concept of RNA γ-interaction structures. Note that the genus of γ-interaction structures is not fixed. For the simplest class, the 0-interaction structures, we already have an unambiguous grammar (Andersen et al., 2012) (Fig. 1). While genus zero structures are exactly RNA secondary structures that only contain noncrossing arcs, genus zero interaction structures exhibit crossings (Fig. 1). Such cross-serial interactions, however, can only occur for exterior arcs, that is, arcs that connect the two backbones.
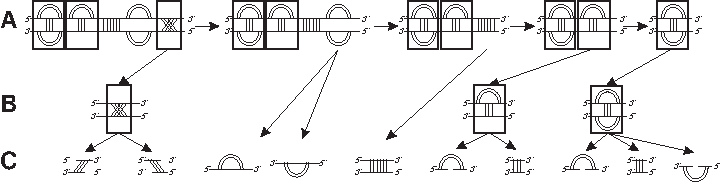
An unambiguous grammar of 0-interaction structures; see Andersen et al. (2012) for details.
The idea of topological RNA interaction structures is a topic of considerable practical interest, since RNA interaction structures that do not belong to the AP structure exist. For instance, the integral RNA (hTER) of the human telomerase ribonucleoprotein has a conserved secondary structure that contains a potential pseudoknot (Ly et al., 2003). There is evidence that the two conserved complementary sequences of one stem of the hTER pseudoknot domain can pair intermolecularly in vitro, and that formation of this stem as part of a novel “transpseudoknot” is required for the telomerase to be active in its dimeric form (Fig. 2).

Can the topological genus really measure the complexity of an RNA structure? An RNA structure is mostly determined by geometric and steric constraints incurred by both canonical and noncanonical base pairs. Secondary structure consists of mainly canonical base pairs between nucleotides offering a scaffold for the tertiary structure, where noncanonical interaction motifs are central. Topological genus provides a natural filtration of RNA pseudoknot structures that is of some importance: for instance, 97% of all RNA pseudoknot structures in databases are composed of irreducible motives of genus one. How then does topology enter the picture, and is topology a phenomenon restricted to pseudoknotted RNA structures? One of the two main approaches to RNA secondary structure folding is to consider and minimize loop energies. Closer inspection reveals that these loops are exactly the topological boundary components. These, in turn, are directly coupled to topological genus via Euler's characteristic, Equation (2.1), described in detail in Section 2. Hence topological genus is an phenomenon that concerns loops in any RNA structure as we have already an implicit appearance of topological genus in RNA secondary structures, that is, the RNA structures of genus zero. However, the genus alone is not the sole discriminator; namely, the finite set of motives appearing for a fixed genus are in fact cell decompositions of a topological surface. In other words, geometry enters the picture in terms of the moduli space of the surface of genus g. As for the noncanonical base pairs that are central for the formation of the RNA tertiary structure, the topological genus and refinements provides the framework for the formulation of noncanonical interactions.
Our main result is the computation of the generating function of γ-interaction structures, Theorem 5. To prove this, we employ γ-structures over one backbone without isolated points (γ-matchings) as stepping stones. Their generating function,
where the
One immediate implication is, that the generating function is algebraic. This implies that the coefficients satisfy polynomial recursions. The existence of the latter is a prerequisite for constructive, algorithmic recursions, which are crucial for fast folding of interaction structures. It may be worth pointing out that the key lies in the derivation of the generating functions. As will become transparent below, there is inherent modularity, which manifests in the symbolic method by which the functions are constructed. The topological interaction structures are controlled by “finite” information in form of specific polynomials, which satisfy explicit recursions.
2. Background And Notations
2.1. Diagrams
A diagram over • a vertex could only incident to at most one A-arc and at most two B-arcs; • any B-arc only exists between consecutive vertices i and i + 1, where
We note here, given an A-arc a and a B-arc b, even though they connect exactly the same pair of vertices, a and b are viewed as different edges instead of multiple edges in the labeled graph. Each diagram is represented by drawing its vertices in increasing order from left to right, together with B-arcs in a horizontal line and A-arcs in the upper half-plane.
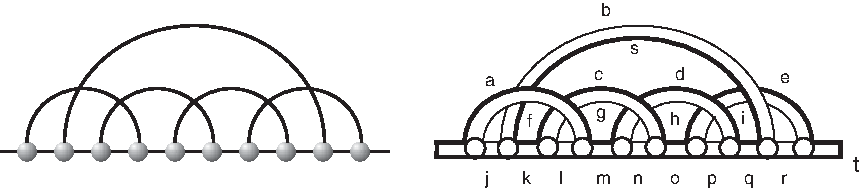
Given a diagram D, consider the edge-induced subgraph of its B-arcs; we call each of its components a backbone. If in total there are b backbones in D, then we say D is a diagram over b backbones. Furthermore, we call the interval [i, i + 1] is a gap interval if there exists a pair of subsequent backbones B1 and B2 such that i(i + 1) is the rightmost (leftmost) vertex of B1(B2). The vertex i is referred to as a cut vertex. There are of course exactly (b − 1) such cut vertices.
A vertex i is isolated if it is not incident to any A-arc. A diagram is connected if it is connected as a graph (i.e., employing A-arcs as well as B-arcs). A diagram that does not contain any isolated vertices is called a matching (Fig. 3).
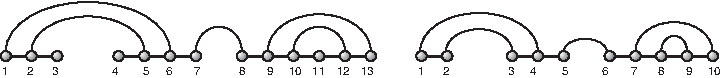
LHS: a diagram over [13] with set of A-arcs {(1, 6), (2, 5), (7, 8), (9, 13), (10, 12)} and set of B-arcs that partition into three backbones: {(1, 2), (2, 3)}, {(4, 5), (5, 6), (6, 7)}, and {(8, 9), (9, 10), (10, 11), (11, 12), (12, 13)}. Here, 3 and 7 are the cut and 3, 4, 11 are isolated vertices. RHS: a matching derived by removing the isolated vertices of the left diagram and relabeling the vertices.
We shall refer to an A-arc simply as an “arc.” Given an arc (i, j), the length of (i, j) is defined by j − i. In particular, when j = i + 1, we refer the arc as a 1-arc. We distinguish exterior and interior arcs, where the former connect a pair of vertices from different backbones and the latter are within a specific one. Diagrams over multiple backbones without exterior arcs are simply disjoint unions of diagrams over one backbone.
An interior stack of length τ on
Furthermore, an interior stack s is τ-canonical if s is of length at least τ. Exterior stacks on [i, j] (SEi,j) and τ-canonical exterior stacks,
Any of these 2(k − 1) intervals is referred to as a P-interval. Note that P-intervals do only emerge in stacks. If P-intervals emerge, they are always of the form [i, i + 1]. Any interval of the form [i, i + 1] other than a gap or P-interval is called a σ interval. Clearly, a diagram over [n] contains (n − 1) intervals of the form [i, i + 1], and we distinguish the three types: gap intervals, P-intervals, and σ-intervals (Fig. 4). Note that by construction, the sets of gap intervals, P-intervals, and σ-intervals are disjoint.
Stacks and intervals: gap intervals, σ-intervals, and P-intervals labeled by
Let ≺ be the partial order on arcs given by (i,j) ≺ (i′, j′) if i′ ≤ i and j ≤ j′. Any diagram has a unique set of maximal arcs (with respect to the partial order), (Fig. 5).
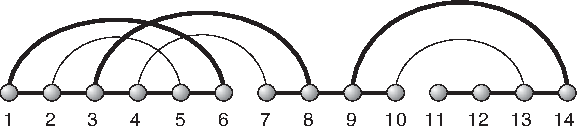
Maximal arcs: the maximal arcs (1, 6), (3, 8), and (9, 14) of a diagram of [14] (bold).
The vertices and arcs of a diagram correspond to nucleotides and base pairs, respectively. For a diagram over b backbones, the leftmost vertex of each backbone denotes the 5′ end of the RNA sequence, while the rightmost vertex denotes the 3′ end. The particular case of b = 2 is referred to as RNA interaction structures (Huang et al., 2009, 2010).
Diagrams over one and two backbones are related by •-product: given two diagrams over two backbones, E1 and E2, we can insert E2 into the gap of E1 by concatenating the backbones R2 and R1 and S1 and S2 preserving orientation and relabeling (Fig. 6). This composition is again a diagram over two backbones denoted by E1 • E2. It is straightforward to see that • is an associative product with units given by the diagram over two empty backbones. The product • is not commutative.
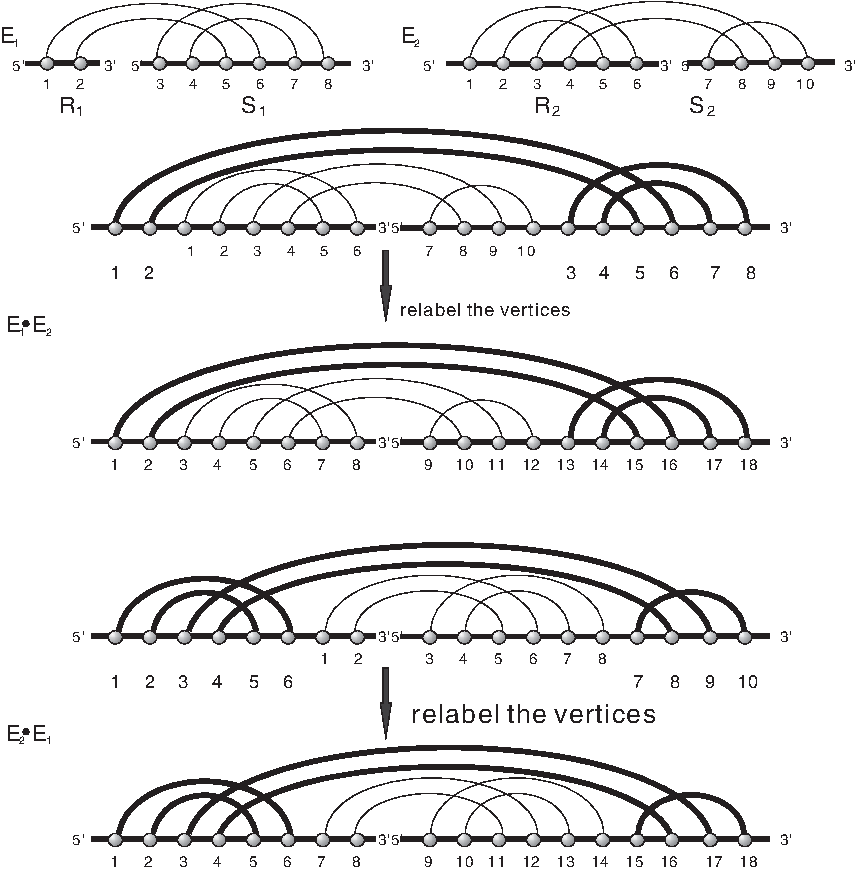
Mapping a diagram over two backbones into a diagram over one backbone by the product •.
2.2. From diagrams to topological surfaces
One approach for deriving meaningful filtration of RNA structure is to pass from diagrams to topological surfaces (Massey, 1967). It is natural to make this transition from combinatorics to topology via fatgraphs (Penner et al., 2010; Penner, 2011). A fatgraph
Computing the number of boundary components. The diagram contains 5+9 edges and 10 vertices. We follow the alternating paths described in the text and observe that there are exactly two boundary components (bold and thin). The genus of the diagram is given by
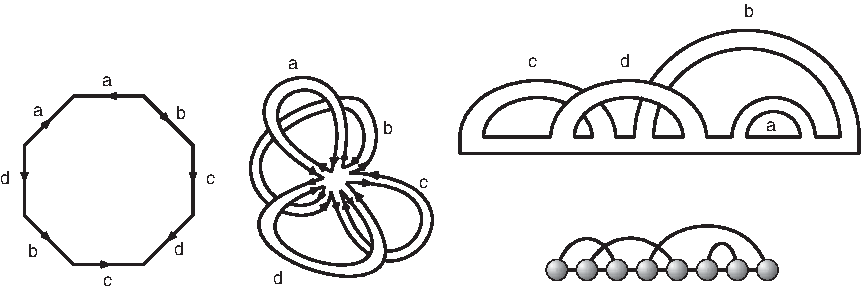
Case study for a diagram over one backbone: equivalence between a pairwise identification of the sides of an octagon (left), the surface F(
where n denotes the number of arcs.
For an RNA diagram, we may draw a representation as usual so that the backbone is a horizontal line oriented from left to right, and the arcs lie in the upper half-plane. This determines a unique fattening on any diagram (cf. the leftmost two panels in Fig. 9 for the fatgraph and its corresponding surface). Each boundary component of F(
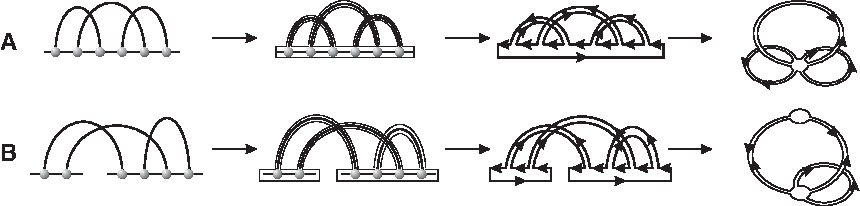
This backbone collapse preserves orientation, Euler characteristic, and genus by construction. It is reversible by inflating each vertex to form a backbone. Using the collapsed fatgraph representation, we see that for a connected diagram over [n] with b backbones, the genus g of the surface (with boundary) is determined by the number n of arcs as well as the number r of boundary components, namely, 2 − 2g− r = |V (G)| − |E(G)| = b − n (cf. Fig. 9), in which v and e are the cardinalities of the vertex set and edge set in the diagram, respectively.
2.3. Shadows
A shadow is a diagram with no noncrossing arcs or isolated vertices in which each stack has length one. The shadow of a diagram is obtained by removing all noncrossing arcs, deleting all isolated vertices, and collapsing each induced stack to a single arc (cf. Fig. 10). Note that projecting into the shadow does not affect genus. In case there are no crossing arcs in a diagram X, then the shadow of X becomes an empty diagram on the same number of backbones as X as in Figure 10C. By definition, any empty backbone contributes one boundary component. For example, for a diagram X over b backbones that contains no crossing arcs, the shadow of X is a sequence of b empty backbones with b boundary components.
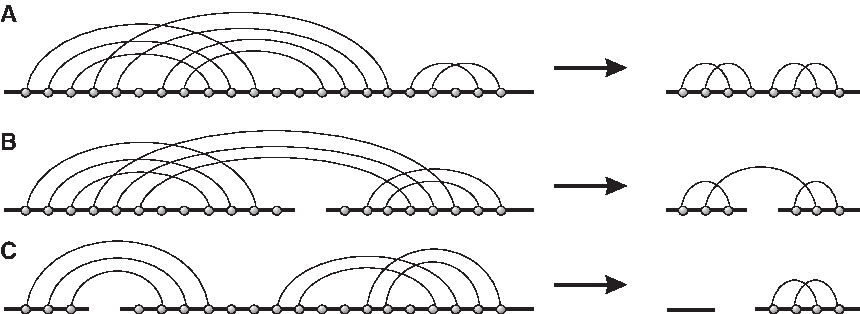
Shadows:
Theorem 1
(Andersen et al., 2012) A shadow over two backbones has the following properties:
Proof. See Appendix A-1. ■
2.4. Irreducibility
A diagram E over b backbones is called irreducible if it is connected and for any two arcs, α1,αk contained in E, there exists a sequence of arcs
such that (αi, αi+1) are crossing. An irreducible shadow is simply a diagram that is a shadow as well as an irreducible diagram. As proved in Andersen et al. (2012), there is the following corollary of Theorem 1.
Corollary 1
An irreducible shadow having genus g = 0 over two backbones contains at least 2 and at most 4 arcs, and for 2 ≤ ℓ ≤ 4, there exists an irreducible shadow of genus g = 0 over two backbones having exactly ℓ arcs. An irreducible shadow having genus g≥1 has the following properties:
Proof. See Appendix A-2. ■
Let X be a diagram. We call S′ an irreducible shadow of X (irreducible X-shadow) if S′ is an irreducible shadow and any arc in S′ is contained in X. S′ is a (g, b, m)-shadow if S′ is a diagram over b backbones having genus g and m arcs. The set of irreducible (g, b, m)-shadows is denoted by
According to Theorem 1, the generating function
We display the seven irreducible shadows over two backbones of genus 0 in Figure 11.
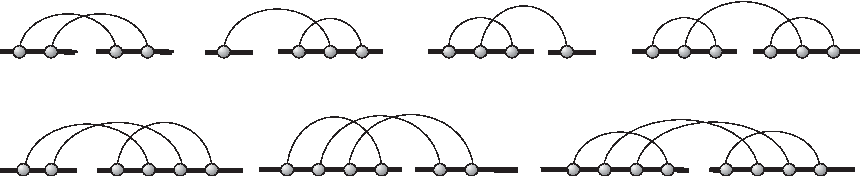
There are exactly seven (3+3+1) irreducible shadows over two backbones of genus 0, in which there are 3/3/1 irreducible shadows with 2/3/4 arcs that correspond to the monomials 3u2, 3u3, and u4 of
2.5. γ-Structures
A diagram is a γ-structure if it is connected and all its irreducible shadows have genus at most γ. A γ-matching is a γ-structure as well as a matching. The combinatorial class of γ-matchings over one backbone is denoted by
Theorem 2
Let
Furthermore, Equation (2.2) determines
in which k is some positive constant and ρ−1≈8.28425.
The combinatorial classes of γ-matchings over two backbones is denoted by
2.6. Symbolic notations
In the following, given a particular set of diagrams
3. γ-Matchings Over Two Backbones
In this section, we compute the generating function
Let
Lemma 1
Let
Proof. Since
When ℓ = 0, it is obvious that
In case of ℓ > 0, set s⋆ to denote the particular
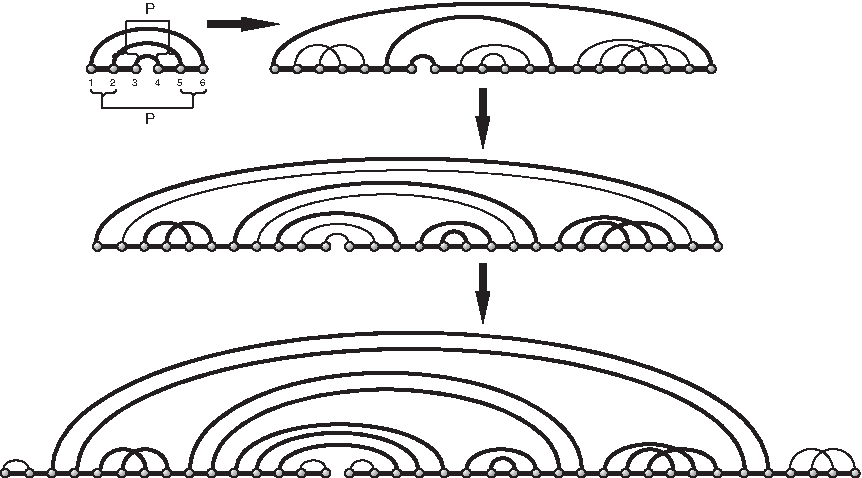
The construction of
(I) Insert nonempty γ-matchings into at least one of the two intervals of each pair of intervals ([i, i + 1], [2ℓ − i, 2ℓ − i + 1]). Since there are ℓ − 1 pairs of such intervals in s⋆, we obtain the class
(II) inflate each exterior arc in the derived matching into an exterior stack. Since there are in total ℓ exterior arcs after Step (I), we derive the class
(III) concatenate each end of the (two) backbones with a γ-matching (possible empty),
Accordingly, we derive
Theorem 3
The generating function of γ-matchings over two backbones satisfies
Proof. To calculate
Let
In case of t = 0,
Next, the calculation of
(I.1) Insert
(I.3) Since, by construction, there is
(II) Secondly, we observe that (3.3) depends on only the number of arcs in φ instead of their particular configuration or the particular ordering of the arcs in φ. Thus, for any other fixed irreducible shadow ϱ of genus g over two backbones having m arcs, we have
and consequently
Note that the summation over m is indeed finite (see Theorem 1).
In the case of t > 1, we proceed by inductively constructing
Set ϱ to be an irreducible shadow in
Since Equation (3.6) depends only on the number of arcs in ϱ, there is
In view of Equation (3.5), we obtain
Together with Equation (3.4), we derive for t ≥ 1
In view of
Theorem 4
Let k0 and k1 be some positive constants. The coefficients of
where
Proof. Suppose first γ = 0. Since
in which,
Pringsheim's theorem guarantees the existence of some real dominant singularity of
where k0 is some positive constant. In Figure 13, we showcase the quality of the asymptotic formula for γ = 0.
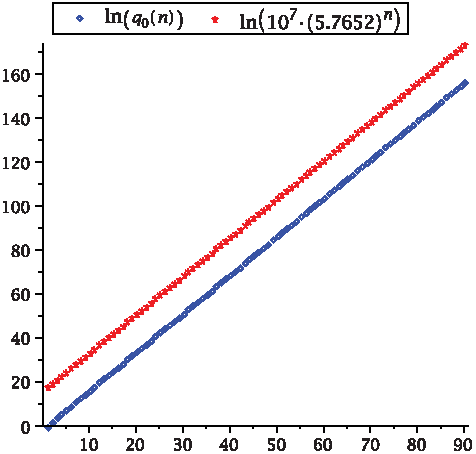
We compare the numbers of
In the case of γ = 1, let δ1 denote the dominant real singularity of
whose existence is implied by Pfringsheim's theorem. According to Theorem 2 (Han and Reidys, 2012),
We set
Since
To derive
for some c > 0 and consequently
where k1 is some positive constant. This completes the proof of the theorem. ■
4. γ-Interaction Structures
The enumeration of γ-interaction structures is obtained using shapes. A matching X is a shape if each stack in X is exactly length one. Given an arbitrary matching s, its shape is obtained by iteratively collapsing each stack to a single arc and then removing any isolated vertices from the obtained diagram as illustrated in Figure 14.
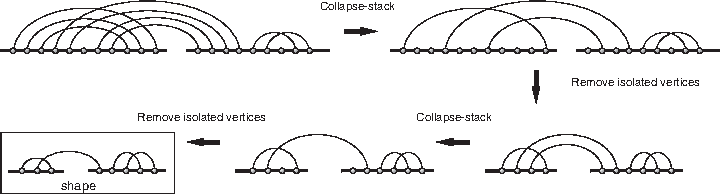
From a diagram to its shape.
Let
Lemma 2
For any γ ≥ 0, we have
Proof. First we claim that
Consider the number of all possible
or equivalently the PDE
which is thus satisfied by
On the other hand,
is also a solution of Equation (4.3), which specializes to
To complete the proof of Equation (4.1), we will define a projection
which projects an arbitrary
Let us consider a fixed γ-shape, λ, having s arcs, of which t are 1-arcs and the generating function
which shows that
Setting
as required. ■
Using symbolic enumeration we can conclude from Lemma 2:
Lemma 3
Let λ be a fixed γ-shape with s ≥ 1 arcs and m ≥ 0 1-arcs. Then the generating function of τ-canonical γ-diagrams containing no 1-arc that have shape λ is given by
In particular,
Proof. Given λ, we can construct an arbitrary τ-canonical γ-diagram containing no 1-arc that has shape λ by the following steps:
• We start from the diagram λ, insert at most one isolated vertex after each given vertices • We insert at most one isolated vertex before the first vertex of the backbones. The obtained combinatorial class is symbolically given by • We insert exactly one isolated vertex after each vertex j such that (j, j + 1) forms an 1-arc in λ. The obtained combinatorial class is symbolically given by • We inflate each arc into a stack of length t ≥ 0. In the case of t > 1, assuming • We inflate each arc in the obtained structure into a stack of length at least τ. The obtained combinatorial class • We inflate each isolated vertex into a sequence of isolated vertices of length at least one. The obtained combinatorial class is symbolically obtained from
Our main result about enumerating τ-canonical γ-interaction structures follows.
Theorem 5
Suppose γ ≥ 0 and τ ≥ 1 and let
In particular, for γ = 0 we have
and for γ = 1 we have
for some constants k1, k2, where
Proof. According to Lemma 3 and
Furthermore, Equation (4.5) follows by substituting
Clearly
5. Discussion
Let us have a look at the impact of this theoretical analysis for biology. While Theorem 3 and Lemma 1 are abstract statements about generating functions, their key insight lies in the recursive construction of genus-filtered interaction structures. Tantamount to the symbolic construction presented here is an algorithmic construction of γ-interaction structures. That is, the mathematical construction has a direct algorithmic translation.
In view of Theorem 2, which specifies the blocks that appear in such structures, we have arrived at the following situation: a genus-filtered RNA interaction structure can only be composed by finitely many building blocks, and Theorem 3 and Theorem 5 show directly how to build recursively any such structure from them. This has direct algorithmic implications: for fixed γ, γ-interaction structures can be folded in polynomial time. Here, Theorem 4 shows that γ-interaction structures are distinctively different from the γ-structures introduced in Han and Reidys (2012). Most prominently, they have, due to restricted ways of nesting (see Theorem 3) not only a significantly lower exponential growth rate but also no genus-dependent subexponential factors. The key difference between γ-structures and γ-interaction structures is their (way of) nesting: there is only one way (which gives rise to the associative product “•”, see Section 2) to nest two γ-interaction structures. As is apparent from Theorem 3, one needs γ-structures in order to build γ-interaction structures and that building process leads to a distinctively different class of RNA structures.
The mere fact that there are only finitely many building blocks has profound algorithmic impact. Namely, it is now possible to devise algorithms that can attribute specific energies to the associated loops, avoiding any generic crossing penalties typical for RNA pseudoknot folding algorithms (Reidys et al., 2011). This has already led to an increased specificity and PPV in the context of folding γ-structures over one backbone (Reidys et al., 2011).
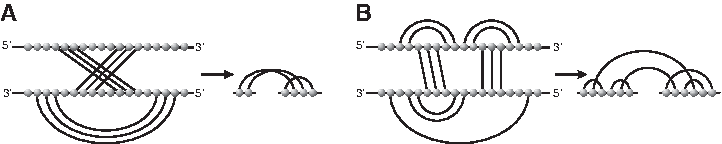
We strongly suspect that such complex interactions are common among the many thousands of long noncoding RNAs, which in the last years have been discovered as important regulatory molecules. It is crucial, therefore, to better understand the theoretical possibilities and to systematically investigate how more complex classes of interaction structure can be addressed algorithmically. In Figure 15, we show that 0-interaction structures are different from AP-structures (Pervouchine, 2004; Alkan et al., 2006).
Let us come back to the recursive constructions that result from algebraic-generating functions. We argued above that the derivation of bivariate algebraic equations as Equation (3.2) is crucial for obtaining recursions for computing shadows of genus g from those of smaller genera. Similar to the Zagier-Harer generating function (Zagier, 1995), the derivation of recursions for the polynomials
Furthermore, this line of work has led to a bijection between genus zero interaction structures and certain pairs of plane trees with some distinguished vertices (Han, 2012). This bijection is new and its algorithmic implications are certainly not fully understood. However, we expect profound impact and novel insights on how to fold interaction structures faster. One immediate implication concerns the parallelization of the interaction structure folding.
In this article, we offer a general mathematical foundation for the nucleic acid interaction problem. This framework is instrumental for devising algorithms for RNA–RNA interaction structures that employ the combinatorial/topological structure of the classification of interaction structures.
6. Appendix
A-1: Proof of Theorem 1
Proof. First, we begin by claiming an observation about shadows over one backbone from Reidys et al. (2011) as follows.
Shadows of genus g ≥ 1 over one backbone have the following properties:
To prove this claim, first note that if there is more than one boundary component, then there must be an arc with different boundary components on its two sides, and removing this arc decreases r by exactly one while preserving g since the number of arcs is given by n = 2g + r − 1. Furthermore, if there are νℓ boundary components of length ℓ in the polygonal model, then 2n = ∑ ℓ ℓν ℓ since each side of each arc is traversed once by the boundary. For a shadow, ν1 = 0 by definition, and ν2 ≤ 1 as one sees directly. It therefore follows that 2n = ∑ ℓ ℓν ℓ ≥ 3(r − 1) + 2, so 2n = 4g + 2r − 2 ≥ 3r − 1, i.e., 4g − 1 ≥ r. Thus, we have n = 2g + (4g − 1) − 1 = 6g − 2, that is, any shadow can contain at most 6g − 2 arcs. The lower bound 2g follows directly from n = 2g + r − 1 since r ≥ 1.
Let S2g be a shadow containing 2g mutually crossing arcs, that is, each arc crosses any of the remaining (2g − 1) arcs. S2g has genus g and contains a unique boundary component of length 4g, that is, traversing 4g nonbackbone arcs counted with multiplicity. We construct a new shadow S2g+1 of genus g containing 2g + 1 arcs by inserting an arc crossing into S2g from the 5′ end of S2g such that the boundary component in S2g splits into one boundary component of length 3 and another of length 4g + 2 − 3 = 4g − 1. The latter becomes the first boundary component of S2g+1. The newly inserted arc is, by construction, crossing, splitting a boundary component, and preserving genus. We now prove the assertion by induction of the number of inserted arcs. By the induction hypothesis, there exists a shadow S2g+i of genus g having 2g + i arcs, whose first boundary component has length 4g − i. Again, we insert a crossing arc as described above, thereby splitting the first boundary component into one of length 3 and the other of length (4g − (i + 1)). After i = 4g − 2 such insertions, we arrive at a shadow whose first boundary component has length 2 while all other boundary components have length 3. Accordingly, there exists a set
We next claim that any shadow of genus g over two backbones can be obtained by cutting the backbone of a shadow over one backbone having either genus g or g + 1. To see this, suppose we are given a shadow of genus g, having r boundary components and n arcs so that 2 − 2g − r = b − n, that is, g = (2 + n − r −b)/2, where b = 1. Cutting the backbone then either splits a boundary component or merges two distinct boundary components. Since cutting does not affect arcs and increases the number of backbones by one, we have the resulting genus
as was claimed. We next observe that a shadow of genus g = 0 over two backbones has at least two arcs, while the maximum number of arcs contained in such a shadow is given by 6(0 + 1) − 2 = 4. For g ≥ 1, it is impossible to cut a shadow of genus g having 2g arcs and keep the genus. Thus, the shadow of genus g over two backbones has at least 2g + 1 arcs. We can always map an arbitrary shadow over two backbones of genus g via α into a shadow over one backbone, whence the assertion. The first claim guarantees that there are only finitely many such shadows, and the theorem follows. ■
A-2: Proof of Corollary 1
Proof. Part (a) follows directly from the first claim in A-1, and for (b), the shadows
Footnotes
Author Disclosure Statement
No competing financial interests exist.