
Abstract
Abstract
In protein engineering, useful information may be gained from systematically generating and screening all single-point mutants of a given protein. We model and analyze an iterative two-stage procedure to generate all these mutants. At each position, L variants are generated in the first stage via saturation mutagenesis and are sequenced. In the second stage, the missing variants (out of the 19 possible single-point substitutions) are produced via site-directed mutagenesis. We study the economic tradeoff associated with varying L, and derive its optimal value given the experimental parameters.
1. Introduction
One way to generate all single-point mutants of a protein is by a massive use of site-directed mutagenesis, in which each mutant is generated individually in a separate reaction. However, the total cost of such a large number of reactions is currently still prohibitive. A more economical alternative is an iterative two-stage procedure, which is to be repeated at each position: (1) saturation mutagenesis is applied to construct L variants, which are all sequenced and observed for which of the 19 substitutions are present; this stage will be referred to as the randomization stage. (2) Site-directed mutagenesis is used to construct individually all the missing substitutions; this stage will be referred to as the site-directed mutagenesis stage.
The main decision variable in this process is L, the number of variants generated and sequenced in the randomization stage. Small L means lower sequencing cost in the randomization stage, yet large L means that more distinct sequences (out of the possible 19) will be generated in the randomization stage, and hence fewer variants will have to be individually generated in the site-directed mutagenesis stage, lowering the cost of this stage. The goal of this work is to establish a methodology for balancing these two conflicting requirements, and thus to optimize the process. Given the various experimental parameters (costs, yield, protein sequence, randomization scheme), one needs to choose optimally L so as to minimize the expected overall cost of the experiment. We emphasize that this work is concerned only with generating the mutant library, and not with the subsequent screening.
2. Methods
Let M be the number of positions to be scanned. These positions may be all of the protein's positions (as in the lipase A experiment, where M = 181), or some predetermined subset thereof. We denote by
We first focus on the cost associated with a single position out of the M to be scanned. This cost depends on the wild-type amino acid at that position, so for position i, we denote it by Ci(L). We assume that the QuikChange protocol is used, so that the per-position cost of the randomization stage is 2cprimer + cfixed + Lcseq (recall that the QuikChange protocol requires two primers—one forward and one backward). The cost of the site-directed mutagenesis stage is Ki,L(2cprimer + cfixed + cseq), where Ki,L is the random variable denoting the number of missing variants that need to be produced individually at position i in the site-directed mutagenesis stage, given that L variants were generated in the randomization stage. The distribution of Ki,L depends also on the randomization scheme (see below), but we suppress this dependence in the notation. The total cost of scanning position i is therefore
The overall cost of the experiment is then
Let na be the number of positions in which the wild-type amino acid is a (so that
To analyze this optimization problem, we turn to study the distribution of Ka,L. Out of the L variants generated in the randomization stage at some given position, let Xa be the number of variants that resulted in amino acid a, and let Xstp be the number of variants which resulted in a premature stop codon (so that
The result now follows from the elementary identity Var(X) = E(X2)−E2(X) and from proposition 1. ■
More generally, it can be shown that the nth moment of Ka,L is
where
Using proposition 1, the optimization problem in Equation (2) can be now written more explicitly as
From proposition 2, the variance of the total cost of the experiment, C(L), is given by
The second derivative is
which is positive for each L ≥ 0, and hence E[C(L)] is convex in L. ■
When viewed as a function of a continuous variable, as we did in the analysis so far, the objective function therefore possesses a unique optimal minimizer L*, which is either the boundary point L = 0, or the solution of the equation (d/dL)E[C(L)] = 0. Because of the structure of the objective function E[C(L)], one cannot solve the latter equation analytically. However, numerical methods for finding L* such as the Newton method are not required, since in practice L is an integer, so one can use, for example, simple binary search across a large enough set of consecutive integers, to find the integer minimizer L*. Note that, in principle, it may happen that there are two integer minimizers, which will be two consecutive integers flanking the real-valued minimizer of the objective function, when viewed as a continuous function of L.
2.1 Other randomization schemes
The cost function used in Equation (1) corresponds to standard QuikChange randomization, in which a single pair of degenerate primers is used. This randomization approach encompasses several widely used schemes, known as NNN, NNK, NNS, and NNB (Chronopoulou and Labrou, 2011). Recently, two other randomization schemes have been proposed: Kille et al. (2013) presented their so-called “22c-trick,” which uses a mixture of three primer pairs—NDT, VHG, and TGG, at 12:9:1 molar ratio. This randomization scheme is more efficient than each of the four abovementioned single-pair schemes, as it requires smaller libraries to attain the same coverage of sequence space. Similarly, the method of Tang et al. (2012), which uses a mixture of four primer pairs—NDT, VMA, ATG, and TGG, at 12:6:1:1 molar ratio—is even more efficient, at the expense of requiring an additional primer pair. To incorporate either 22c-trick randomization or Tang randomization into the analysis, all that needs to be done is to change the coefficient of the parameter cprimer in Equations (3) and (4) from 2 to either 6 or 8, respectively, and to update the probabilities pa, a ∈ A (in the 22c-trick randomization, pLeu = pVal = 2/22 and pa = 1/22 for a ≠ Leu,Val; in Tang randomization, pa = 1/20 for all a ∈ A).
2.2 Yield
The analysis so far assumed that both the randomization and the site-directed mutagenesis are always successful. In practice this is not always the case, and one determines the yield of the experiment as the probability that the intended mutation is inserted successfully into the gene of interest. Let α ∈ (0, 1] denote the yield of the experiment.
The yield influences the above analysis at both stages of the process. At the randomization stage, the probability pb that the randomization will result in amino acid b is lowered by a factor α. At the site-directed mutagenesis stage, when generating each of the Ki,L missing variants, there is a probability 1−α that sequencing will reveal that the cloning failed, so the mutagenesis will have to be repeated until it is performed successfully. The total cost in Equation (1) therefore changes to
where Ni,j is the number of cloning trials of variant j (out of the Ki,L missing ones) until successful cloning has occurred, in the site-directed mutagenesis stage of position i. From this definition, Ni,j is geometrically distributed, with probability of success α.
Using Wald's equation for the mean of a random sum, the objective function of Equation (3) changes to
The optimal L* can be still easily found using the approach described above.
Using the well known equation for the variance of a random sum, the variance of the total cost (Equation (4)) changes to
where E(Ni,j) = 1/α and Var(Ni,j) = (1−α)/α2 (the mean and variance of a geometric distribution), and
3. Results
In this section, we present the practical consequences of the mathematical results derived in the Methods section.
The left panel of Figure 1 shows the expected overall cost of the experiment as a function of L, the number of variants generated and sequenced in the randomization stage. The black curve corresponds to the ideal case of 100% yield, and the red curve to 80% yield. In both cases it was assumed that randomization was done using NNK degenerate oligonucleotides, and that the cost parameters are cprimer = 8, cfixed = 2, and cseq = 2.8 (these are realistic figures in Euros, at the time of writing this article).
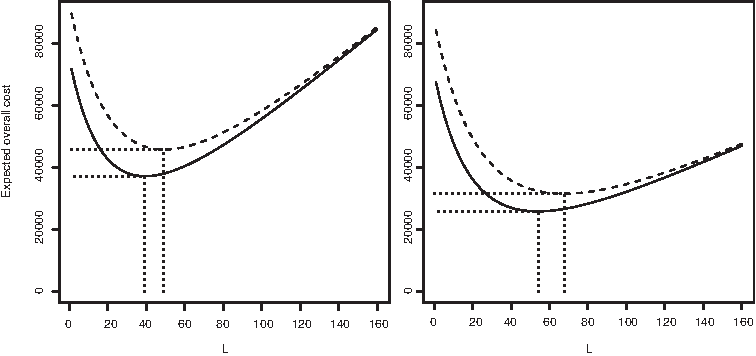
Expected overall cost as a function of L, the number of variants generated and sequenced in the randomization stage. Black curve corresponds to 100% yield and red to 80%. Left: cprimer = 8, cfixed = 2, and cseq = 2.8. Right: cseq lowered to 1.5. All curves refer to NNK randomization and to a protein with M = 181 randomized positions, as in the Bacillus subtilis lipase A experiment.
It can be seen that the optimal L in the case of 100% yield is L* = 39, which correspond to an optimal expected overall cost of E[C(L*)] = 37,272. The cost curve varies considerably near its minimum: Sequencing L = 96 variants in the randomization stage (an entire microplate) would result in an expected overall cost of 54,012, an increase of 45%. As expected, the optimal overall expected cost increases when the yield drops to 80%, to 45,877, which is attained with L* = 49.
Over the past 2 decades, technological advances have kept lowering biotechnological costs considerably, and are expected to continue doing so. The right panel of Figure 1 shows how the results change when the sequencing cost cseq drops from 2.8 to 1.5. With 100% yield, the optimal value of L is L* = 54, which correspond to an optimal expected overall cost of 25,814. Under 80% yield, the figures change to L* = 68 and E[C(L*)] = 31,522.
The influence of the yield on the expected overall cost can be seen in more detail in Figure 2. The left panel of the figure depicts the optimal value of the cost function (the minimal point of the cost curve in Figure 1) as a function of the yield. As expected, the resulting curve tends to infinity as the yield approaches zero. The influence of decreasing the yield on L* is a priori less clear, as lower yield affects adversely both stages of the scanning process. The right panel of Figure 2 shows that not only the optimal overall cost, but also L*, increases as the yield tends to 0.

Left: The optimal expected overall cost, E[C(L)], as a function of the yield. Right: The optimal number of variants generated and sequenced in the randomization stage, L*, as a function of the yield.
Although the distribution of C(L) is certainly not normal, it may be well approximated by a normal distribution having the mean and variance of C(L) (Equations (3) and (4) in the Methods section), as shown in Figure 3. The central limit theorem explains the good fit when M, the number of scanned positions, is large (M = 181 in the left panel), but the fit remains good also when only few positions are scanned (M = 10 in the right panel).

Histogram of 10,000 simulated values of C(L), the total cost of the experiment, with a density plot of a normal distribution having the mean and variance of C(L). Left: M = 181, as in the lipase A study. Right: M = 10. In both cases L = 40, and other parameters are as in the left panel of Figure 1.
All the results presented so far assumed that NNK randomization was used. Figure 4 shows the expected overall cost as a function of L for two additional randomization schemes: the “22c-trick,” and the method of Tang et al. (2012). It can be seen that in both methods, the savings due to the improved efficiency do not compensate for the increase in primer cost.

Expected overall cost as a function of L, the number of variants generated and sequenced in the randomization stage, for three randomization schemes. Left: 100% yield. Right: 80% yield. Other parameters are as in the left panel of Figure 1.
4. Discussion
The goal of this work is to establish a methodology for determining the optimal resource allocation, when scanning all single-point mutants of a protein. Only a small adjustment is needed if one wishes to scan not all 19 substitutions at each position, but rather, some other predetermined number. One can analyze similarly also a process whereby all two-mutant variants of a protein are to be systematically generated.
The cost function we employ is rather simplistic, as it ignores, for example, the cost of the nonexpendable laboratory equipment. Still, it captures the most essential components of the situation: the tradeoff between increased randomization cost and increased site-directed mutagenesis cost, as a result of varying L, the number of variants generated and sequenced in the randomization stage. Neglecting the integrality constraint, the optimal L is the solution of the equation obtained by equating the derivative of the objective function to zero. This solution lends itself to an intuitive interpretation: When increasing L by a unit, the per-position increase in the randomization cost is the derivative of the expression 2cprimer + cfixed + Lcseq, which is exactly cseq, as one more variant will have to be sequenced. At the same time, the expected decrease in the site-directed mutagenesis cost (a decrease that takes place as fewer variants will have to be generated individually) is the derivative of (2cprimer + cfixed + cseq)E(Ka,L). The optimal L is indeed the value that makes these two forces equal to each other, and is therefore found by equating to zero the derivative of their sum, the total expected cost.
The original problem formulation assumed that the same L will be used in the randomization stage for all of the protein's position. However, since each position is dealt with separately, and since the per-position cost Ci(L) depends on the specific wild-type amino acid at that position, one could in principle find and use an optimal L for each position. By doing so the overall expected cost will indeed decrease, but it can be shown mathematically that the decrease will be of only about 1%, a small difference that will most likely get buried in the experimental noise.
Our analysis shows that the yield has a substantial influence on both the optimal cost and on the optimal L required to attain it. We recommend estimating the yield in a small preliminary experiment, and then use the estimate for determining the optimal L in the full-scale experiment.
In the process studied in this work, the genetic diversity created in the randomization stage was generated via saturation mutagenesis. Other alternatives are random mutagenesis techniques such as error-prone PCR (Cadwell and Joyce, 1992) and SeSaM (Wong et al., 2004), which have been used successfully in many mutagenesis experiments. However, upon analyzing mathematically these alternatives, it turns out that even under ideal conditions (zero probability for premature stop codons and complete control over the mutation spectrum), the resulting expected overall cost will be considerably higher than that obtained via saturation mutagenesis. The reason for this is that random mutagenesis protocols are whole-protein methods, which invariably produce a large portion of multiple-point mutants, whereas the experimental goal discussed in our work is to generate systematically only single-point mutants. The inevitable additional cost, incurred by generating and sequencing these multiple-point mutants, makes random mutagenesis methods less suitable for our specific goal. Another alternative for creating the desired library would be via a massive use of gene synthesis; however, at the current synthesis prices, this approach will be about an order of magnitude more expensive than the two-stage process studied above.
Footnotes
Acknowledgments
We thank Ulrich Krauss, group “Molecular Biophotonics,” Institute of Molecular Enzyme Technology, for advice and discussions. AF and KEJ gratefully acknowledge support by the German Research Foundation (DFG) within research training group 1166 “Biocatalysis using Non-Conventional Media—BioNoCo”.
Disclosure Statement
No competing financial interests exist.