
Abstract
Abstract
The analysis of the sequence–structure relationship in RNA molecules is not only essential for evolutionary studies but also for concrete applications such as error-correction in next generation sequencing (NGS) technologies. The prohibitive sizes of the mutational and conformational landscapes, combined with the volume of data to process, require efficient algorithms to compute sequence–structure properties. In this article, we address the correction of NGS errors by calculating which mutations most increase the likelihood of a sequence to a given structure and RNA family. We introduce RNApyro, an efficient, linear time and space inside–outside algorithm that computes exact mutational probabilities under secondary structure and evolutionary constraints given as a multiple sequence alignment with a consensus structure. We develop a scoring scheme combining classical stacking base-pair energies to novel isostericity scores and apply our techniques to correct pointwise errors in 5s and 16s rRNA sequences. Our results suggest that RNApyro is a promising algorithm to complement existing tools in the NGS error-correction pipeline.
1. Introduction
For half a century, biological molecules have been studied as proxies to understanding evolution (Zuckerkandl and Pauling, 1965), and because of their fundamental functions and remarkably conserved structures, rRNAs have always been a prime candidate for phylogenetic studies (Olsen et al., 1986; Olsen and Woese, 1993). In recent years, studies such as the Human Microbiome Project (Turnbaugh et al., 2007) benefited from new technologies such as the NGS techniques to sequence as many new organisms as possible and extract an unprecedented flow of new information. Nonetheless, these high-throughput techniques typically have high error rates that make their applications to metagenomics (aka environmental genomics) studies challenging. For instance, pyrosequencing as implemented by Roche's 454 may have an error rate raising up to 10%. Because there is no cloning step, resequencing to increase accuracy is not possible, and it is therefore vital to disentangle noise from true sequence diversity in this type of data (Quince et al., 2009). Errors can be significantly reduced when large multiple sequence alignments with close homologs are available, but in studies of new or not well-known organisms, such information is rather sparse. In particular, it is common that there is not enough similarity to differentiate between the sequencing errors and the natural polymorphisms that we want to observe, often leading to artificially inflated diversity estimates (Kunin et al., 2010). A few techniques have been developed to remedy to this problem (Quinlan et al., 2008; Medvedev et al., 2011), but they do not take into account all the available information. It is therefore essential to develop methods that can exploit any type of signal available to correct errors.
In this article, we introduce RNApyro, a novel algorithm that enables us to calculate precisely mutational probabilities in RNA sequences with a conserved consensus secondary structure. We show how our techniques can exploit the structural information embedded in physics-based energy models, covariance models, and isostericity scales to identify and correct pointwise errors in RNA molecules with conserved secondary structure. In particular, we hypothesize that conserved consensus secondary structures combined with sequence profiles provide information that allows us to identify and fix sequencing errors.
Here, we expand the range of algorithmic techniques previously introduced with the RNAmutants software (Waldispühl et al., 2008; Waldispühl and Ponty, 2011). Instead of exploring the full conformational landscape and sample mutants, we develop an inside–outside algorithm that enables us to explore the complete mutational landscape with a fixed secondary structure and to calculate exactly mutational probability values. In addition to a gain in the numerical precision, this strategy allows us to drastically reduce the computational complexity (
We design a new scoring scheme combining nearest-neighbor models (Turner and Mathews, 2010) to isostericity metrics (Stombaugh et al., 2009). Classical approaches use a Boltzmann distribution whose weights are estimated using a nearest-neighbor energy model (Turner and Mathews, 2010). However, the latter only accounts for canonical and wobble base pairs. As was shown by Leontis and Westhof (2001), the diversity of base pairs observed in tertiary structures is much larger, albeit their energetic contribution remains unknown. To quantify geometrical discrepancies, an isostericity distance has been designed (Stombaugh et al., 2009), increasing as two base pairs geometrically differ from each other in space. Therefore, we incorporate these scores in the Boltzmann weights used by RNApyro.
We illustrate and benchmark our techniques for pointwise error corrections on the 16S and 5S ribosomal RNAs. We choose the latter since it has been extensively used for phylogenetic reconstructions (Hori and Osawa, 1987), and its sequence has been recovered for over 712 species (in the Rfam seed alignment with id RF00001). Using a leave-one-out strategy, we perform random distributed mutations on a sequence. While our methodology is restricted to the correction of pointwise error in structured regions (i.e., with base pairs), we show that RNApyro can successfully extract a signal that can be used to reconstruct the original sequence with an excellent accuracy. This suggests that RNApyro is a promising algorithm to complement existing tools in the NGS error-correction pipeline.
The algorithm and the scoring scheme are presented in Section 2. Details of the implementation and benchmarks are in Section 3. Finally, we discuss future developments and applications in Section 4.
2. Methods
We introduce a probabilistic model, which aims at capturing both the stability of the folded RNA and its affinity toward a predefined 3D conformation. To that purpose, a Boltzmann weighted distribution is assumed, based on a pseudo-energy function E(·), which includes contributions for both the free energy and its putative isostericity toward a multiple sequence alignment. In this model, the probability that the nucleotide at a given position needs to be mutated (i.e., corresponds to a sequencing error) can be computed using a variant of the inside–outside algorithm (Lari and Young, 1990) in time, which scales linearly with the sequence length.
2.1. Probabilistic model
Let Ω be a gap-free RNA alignment sequence and S its associated secondary structure, then any sequence s has probability proportional to its Boltzmann factor
where R is the Boltzmann constant, T the temperature in Kelvin, ES(s) and EI(s, S, Ω) are the free-energy and isostericity contributions respectively (further described below), and
where
is the average isostericity of a base pair in the candidate sequence, compared with the reference alignment. The ISO function uses the Watson-Crick/Watson-Crick cis isostericity matrix computed by Stombaugh et al. (2009). Isostericity scores range between 0 and 9.7, 0 corresponding to a perfect isostericity, and a penalty of 10 is used for missing entries. The isostericity contribution exponentially favors sequences that are likely to adopt a similar local conformation as the sequences contained in the alignment.
2.2. Computing the mutational profile of sequences
Let s be an RNA sequence, S a reference structure, and m ≥ 0 a desired number of mutations. We are interested in the probability that a given position contains a specific nucleotide, over all sequences having at most M mutations from s. Formally, let
Clearly, the number of sequences in
The former, defined in Equations (3) and (4)–(6), is analogous to an inside computation: Considering a given substructure of the input structure, it computes the accumulated contributions of any possible sequences that have suitable Hamming distance within the interval. It is therefore similar to a partition function, that is, the sum of Boltzmann factors over all sequences within [i, j], knowing that position i−1 is composed of nucleotide a (resp. j+1 is b), within m mutations of s.
The latter, defined by Equations (7) and (8)–(11), computes the outside algorithm, the partition function over sequences within m mutations of s outside the interval (restricted to two intervals [0, i] ∪ [j, n- 1]), knowing that flanking inner positions (i+1, j−1) contain nucleotides a and b respectively. A suitable combination of these terms, computed as shown in Equations (13)–(15), gives the total weight of every possible sequence that supports a given base-pair (or unpaired position) and, in turn, the probability of seeing a specific base at a given position.
In other words, either there is no sequence at distance m > 0 of the empty sequence, or the only allowed sequence is the empty sequence (m=0), having energy 0. Since the contributions only depend on base pairs, they do not appear in the initial conditions.
The main recursion considers a general structure S, flanked by two nucleotides (outside the region of interest) x and y, respectively, on the 5′ and 3′ end of the sequence. As illustrated by Figure 1, it is computed, for each subinterval [i, j], by considering one of the three following cases, dependent on the base-pairing status and context of the leftmost position in the sequence/structure:
– Indeed, any suitable sequence is a concatenation of a, possibly mutated, nucleotide a′ at the first position, followed by a sequence over the remaining interval, having m−δa,a′ mutations (accounting for a possible mutation at first position), and having flanking nucleotides a′ and y. – Any sequence generated here consists of two possibly mutated nucleotides a′ and b′, flanking a sequence over the remaining portion. In order for the total distance to sum to m, this portion must feature m−δab,a′b′ additional pointwise mutations. – In other words, if the leftmost position is paired, and the base pair is not stacking onto another base pair, then the only term contributing directly to the energy is the isostericity of the base pair. Admissible sequences for S consist of two paired nucleotides a′ and b′ at positions i and k respectively, flanking a sequence for S′ (over an interval [i+1, k−1]) and followed by a (possibly empty) sequence for S′′ (over [k+1, j]). Since the total number of mutations sums to m, a parameter m′ is introduced to distribute the remaining mutations between the two sequences.
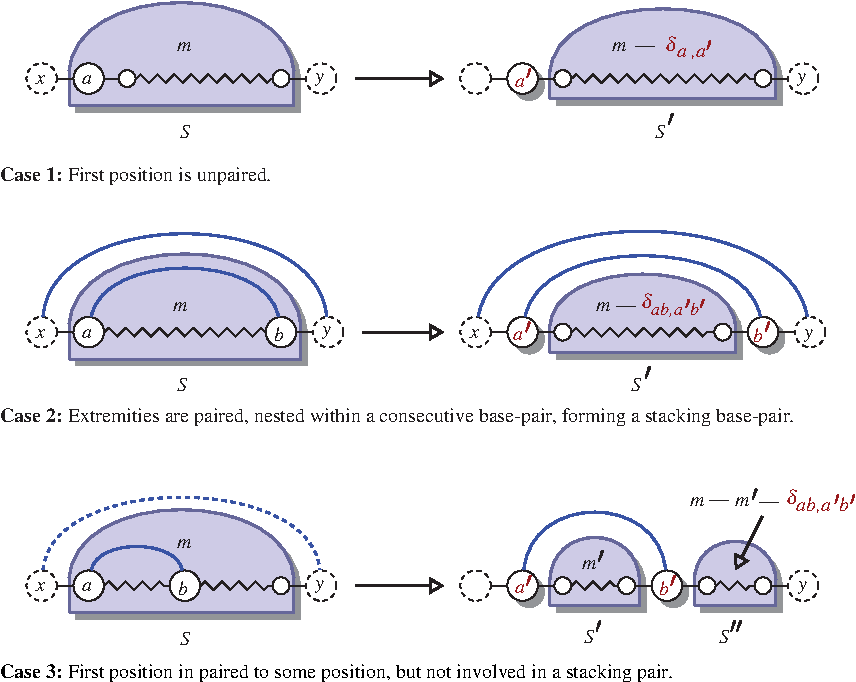
Principle of the inside computation (partition function). Any sequence (mutated) can be decomposed as a sequence preceded by a possibly mutated base (unpaired case), a sequence surrounded by a possibly mutated base-pair (stacking-pair case), or as two sequences segregated by a possibly mutated base-pair (general base-pairing case). In this latter case, mutations must be distributed between subsequences.
where
In general, the main recurrence below works by extending the excluded structure S (covering the ]i, j[ interval) in the leftward direction. As shown in Figure 2, four cases must be considered depending on the base-pairing status and directionality of the next considered position:
– – – –
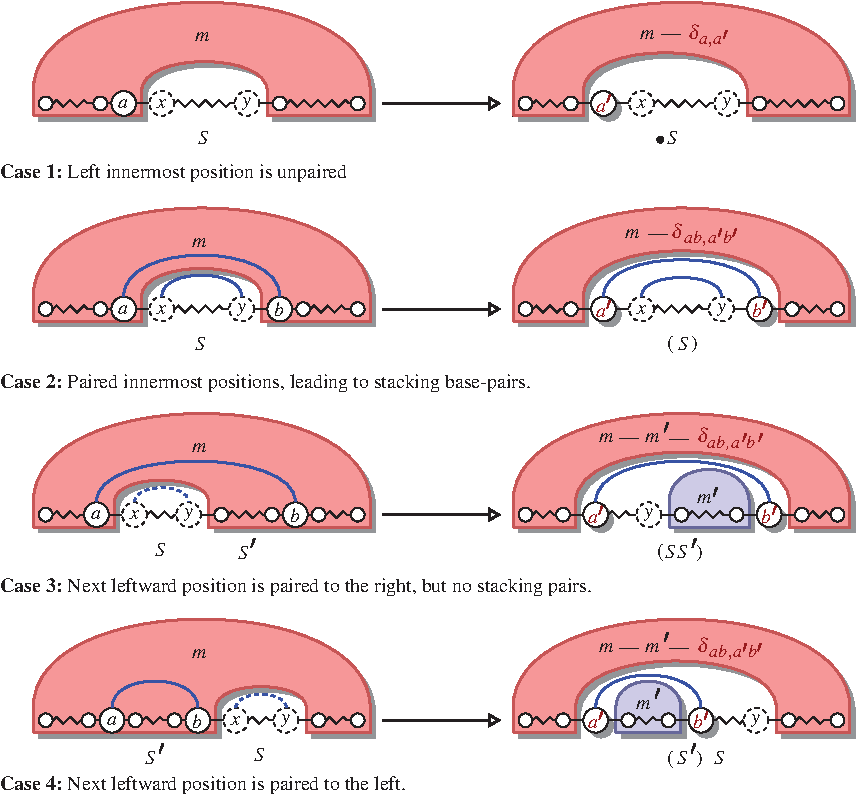
Principle of the outside computation. Note that the outside algorithm uses intermediate results from the inside algorithm; therefore, its efficient implementation requires the precomputation and storage of the inside contributions.
where
To that purpose, one leverages the inside–outside construction as illustrated in Figure 3. Namely, while computing the total contribution of sequences featuring a base – – –

Combining the inside and outside contributions to compute the total Boltzmann weight of all sequences having a given base (case 1) or base pair (cases 2 and 3) at a given position.
2.3. Complexity considerations
Using dynamic programming, Equations (4)–(6) and (8)–(11) can be computed in linear time and memory. Namely, the
In principle, Θ(M · n2) terms, identified by different (S, m) triplets, should be computed. However, a close inspection of the recurrences reveals that the computation can be safely restricted to a subset of intervals (i, j). For instance, the inside algorithm only requires computing intervals [i, j] that do not break any base pair, and whose next position j+1 is either past the end of the sequence or is base-paired prior to i. A similar property holds for the outside computation, following from the linearity of the outside recurrence (i.e., the computation of the outside term for a given excluded structure only relies on the computation of another, strictly larger, structure).
These properties drastically limit the combinatorics of required computations, dropping from Θ(n2) to Θ(n) the number of terms that need to be computed and stored. Consequently the overall complexity of the algorithm is Θ(n · (|Ω|+M2)) arithmetic operations and Θ(n · (|Ω|+M)) memory.
3. Results
3.1. Implementation
The software was implemented in Python2.7 using the mpmath (Johansson et al., 2010) library for arbitrary floating point precision. The source code is freely available online.
The time benchmarks were performed on an AMD Opteron Processor 6278 at 2.4 GHz with cache of 512 KB, (20 cores, and 175 GB ram. Since typical use-cases of RNApyro require efficiency and scalability, we present in Figure 4 typical runtimes required to compute the probabilities for every nucleotide at every position for a vast set of parameters. For those tests, both the multiple sequence alignment and the reference sequence were generated uniformly at random, based on a realistic random secondary structure, generated as described in Levin et al. (2012).
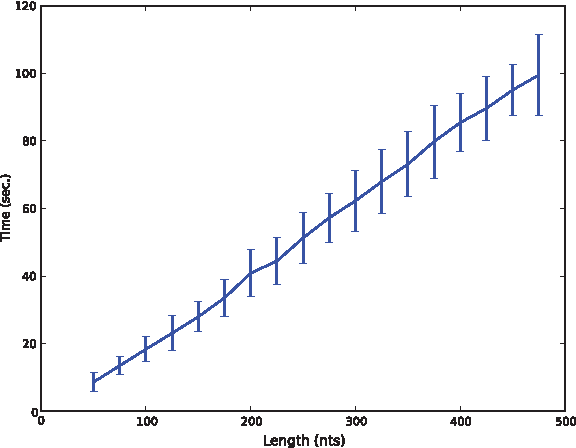
Typical runtimes required by the computation of mutational profiles averaged on 50 random sequences for each length ranging from 100 and 400 nts, while allowing for a maximal number of mutations, M=10. For each sequence, a random multiple sequence alignment was generated, consisting ozf 51 aligned sequences, each compatible with a randomly generated consensus secondary structure.
3.2. Homogenous error correction in 5s rRNAs
To illustrate the potential of our algorithm, we applied our techniques to identify and correct pointwise errors in RNA sequences with conserved secondary structures. More precisely, we used RNApyro to reconstruct 5s rRNA sequences with randomly distributed mutations. This experiment has been designed to suggest further applications to error-corrections in pyrosequencing data.
We built our data set from the 5S rRNA multiple sequence alignment (MSA) available in the Rfam Database 11.0 (Rfam id: RF00001). Since our software does not currently implement gaps (mainly because scoring indels is a challenging issue that cannot be fully addressed in this work), we clustered together the sequences with identical gap locations. From the 54 MSAs without gap produced, we selected the biggest MSA, which contains 130 sequences (out of 712 in the original Rfam MSA). Then, in order to avoid overfitting, we used cd-hit (Li and Godzik, 2006) to remove sequences with more than 80% of sequence similarity. This operation resulted in a data set of 45 sequences.
We designed our benchmark using a leave-one-out strategy. We randomly picked a single sequence from our data set and performed 12 random mutations, corresponding to an error-rate of 10%. We repeated this operation 10 times. The value of β was set to 15 (larger values gave similar results). To estimate the impact on the distribution of the relative weights of energy and isostericity, we used four different values of α=0,0.5,0.8,1.0. Similarly, we also investigated the impact of an under- and overestimate of the number of errors, by setting the presumed number of errors to 50% (6 mutations) and 200% (24 mutations) of their exact number (i.e., 12).
To evaluate our method, we computed a ROC curve representing the performance of a classifier based on the mutational probabilities computed by RNApyro. More specifically, we fixed a threshold
where ω[i] is the nucleotide at position i in the input sequence. We note that, for lower thresholds, multiple nucleotides may be available in C(i) to correct the sequence. Here, we remind the reader that our aim is to estimate the potential of error-correction of RNApyro and not to develop a full-fledged error-correction pipeline, which shall be the subject of further studies. Finally, we progressively varied λ between 0 and 1 to calculate the ROC curve and the area under the curve (AUC). Our results are reported in Figure 5.
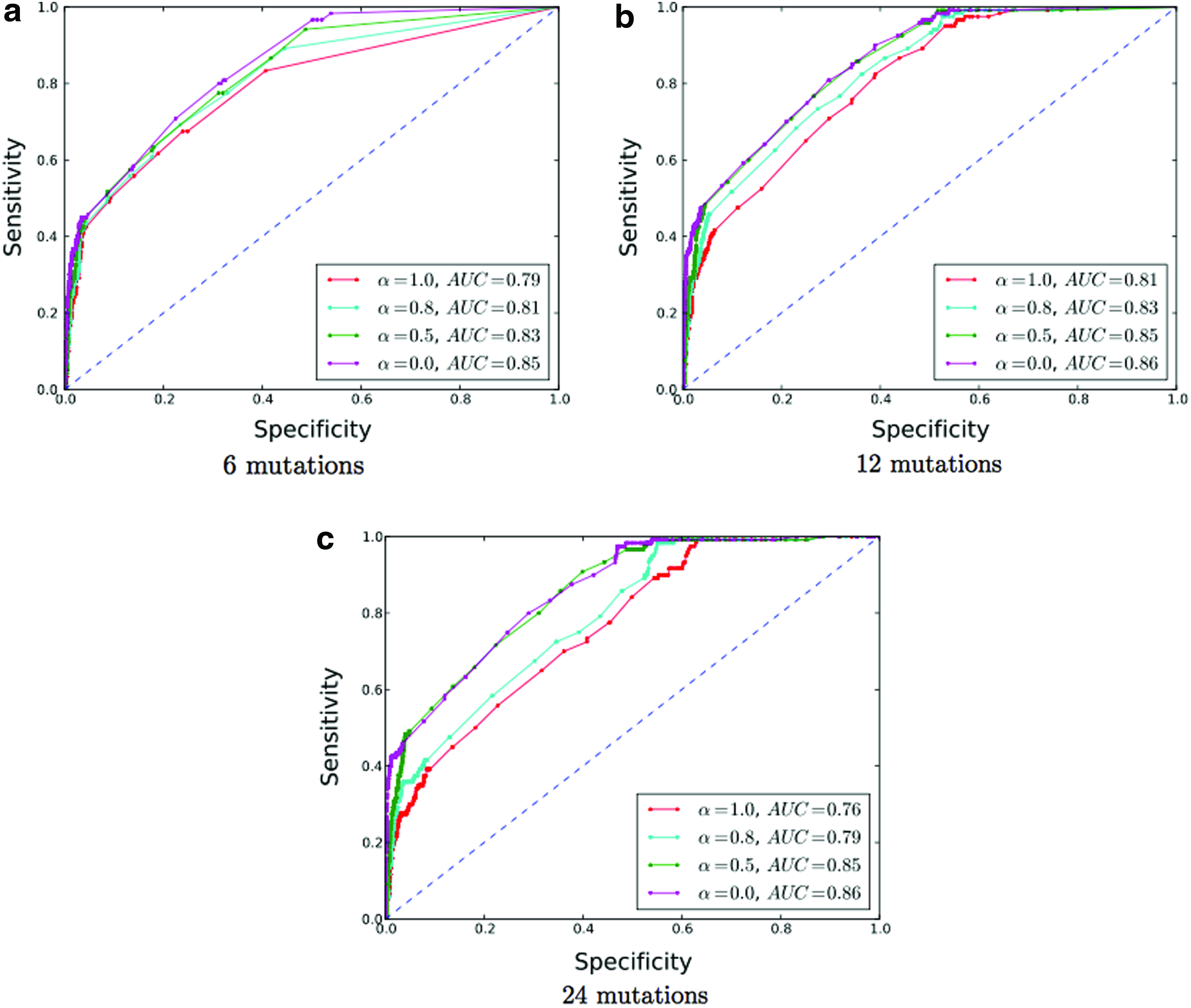
Performance of error-correction. Subfigures show accuracy with underestimated error rates (6 mutations), exact estimates (12 mutations), and overestimates (24 mutations). We also analyze the impact of the parameter α distributing the weights of stacking pair energies versus isostericity scores and use values ranging of α={0,0.5,0.8,1.0}. The AUC is indicated in the legend of the figures. Each individual ROC curve represents the average performance over the 10 experiments.
Our data demonstrates that our algorithm shows interesting potential for error-correction applications. First, the AUC values (up to 0.86) indicate that a signal has been successfully extracted. This result has been achieved with errors in loop regions (i.e., without base-pairing information) and thus suggests that correction rates in structured regions (i.e., base-paired regions) could be even higher. Next, the optimal values of α tend to be close to 0.0. This finding suggests that, at this point, the information issued from the consideration of stacking energies is currently modest. However, specific examples showed improved performance using this energy term. Further studies must be conducted to understand how to make the best use of it. Finally, our algorithm seems robust to the number of presumed mutations. Indeed, good AUC values are achieved even with conservative estimates for the number of errors (cf, 50% of the errors, leading to Fig. 5a), as well as with large values (cf, 200% of the errors in Fig. 5c). It is worth noting that scoring schemes giving a larger weight on the isostericity scores (i.e., for low α values) seem more robust to under- and overestimating the number of errors.
3.3. Correcting Illumina sequencing errors in 16s rRNAs
To complete our benchmark, we turned to the small subunit ribosomal RNA in bacteriae, a molecule which is of particular interest in metagenomics and phylogenetics. Our aim was to get as close as possible to a pyrosequencing context in which reads are produced nonuniformly by an Illumina sequencer, impacting the distribution of errors in the sequence. We chose this setting both because of the popularity of Illumina sequencing in metagenomics, and since the underlying sequencing technique only considers base substitutions (no insertions), the only type of errors currently detected by RNApyro. To that purpose, we used simulated Illumina reads, mapped the reads back to a reference alignment, and ran RNApyro on a consensus sequence derived from the mapped reads, estimating the maximal amount of mutations from both the length of the sequence and the sequencing depth.
We gathered the seed sequences of the bacterial multiple sequence alignment (MSA) retrieved from the RFAM Database 11.0 (Rfam id: RF00177) (Gardner et al., 2011). This alignment is composed of 93 sequences, whose length varies between 1461 and 1568 nucleotides and has an average pairwise sequence identity of 69%. We used the pseudoknot-free version of the consensus secondary structure. A secondary structure for a specific reference sequence was obtained by simply mapping the structure back to sequence, that is, by removing any base pair having at least one partner involved in a gapped position. For similar reasons, we locally excluded from our calculation of the isostericity contribution for a given base pair, described in Section 2.1, any sequence featuring at least a gap on the corresponding positions.
To simulate sequencing errors, we used the next-generation sequencing read simulator ART (Huang et al., 2012). The Illumina technology setting was chosen as the main error mode, generating reads of 75 bps, featuring mostly base substitution errors. Reads were mapped back to the original sequence, and a consensus sequence was determined from the sequencing output by a simple majority vote in the case of multiple coverage. Uncovered regions were simply generated at random. The average rate of errors observed in the final consensus sequence was empirically estimated to represent 2.4%, 0.9%, and 0.6% of the reference sequence for prescribed sequencing coverages of 5-, 10-, and 15-fold respectively.
As in Section 3.2, we evaluated the predictive power of RNApyro-computed mutational profiles using a leave-one-out strategy. We picked a sequence at random from the MSA, sequenced/mutated it as above, and ran RNApyro to establish its mutational profile. In this execution, we used values of β=15 and
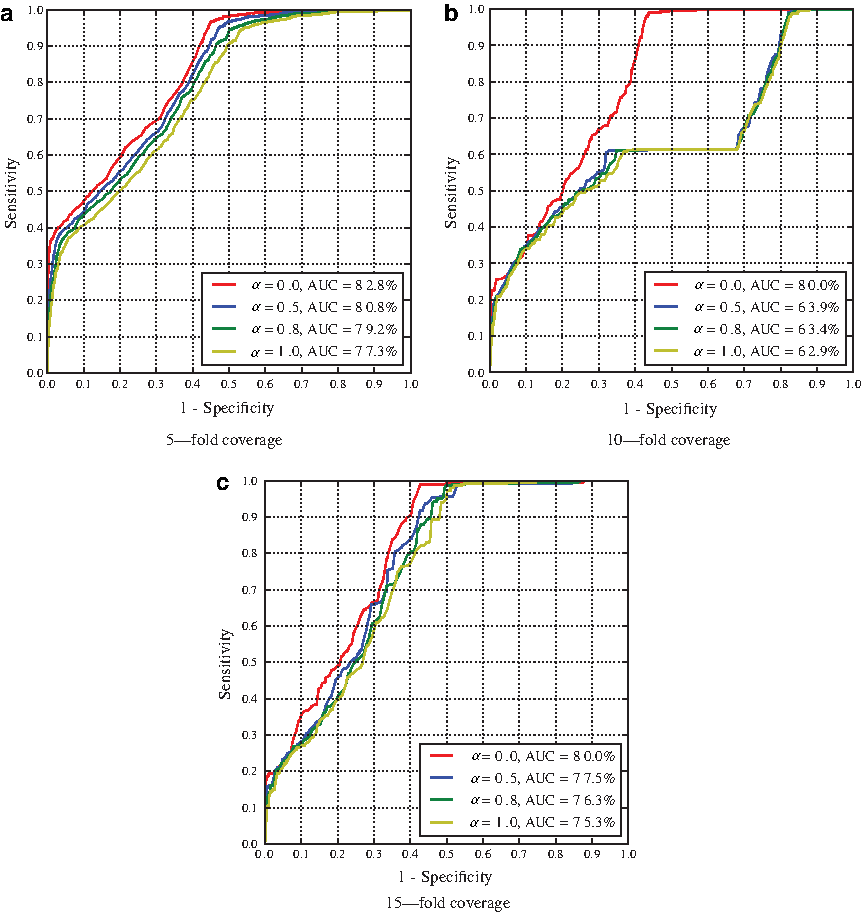
Performance of error-correction over all positions. Subfigures show accuracy when ART fold parameter is set to 5-, 10-, and 15-fold coverage. We also analyze the impact of the parameter α distributing the weights of stacking pair energies versus isostericity scores and use values ranging from α = {0,0.5,0.8,1.0}. The AUC is indicated in the legend of the figures. Each individual ROC curve represents the average performance over 30 experiments.
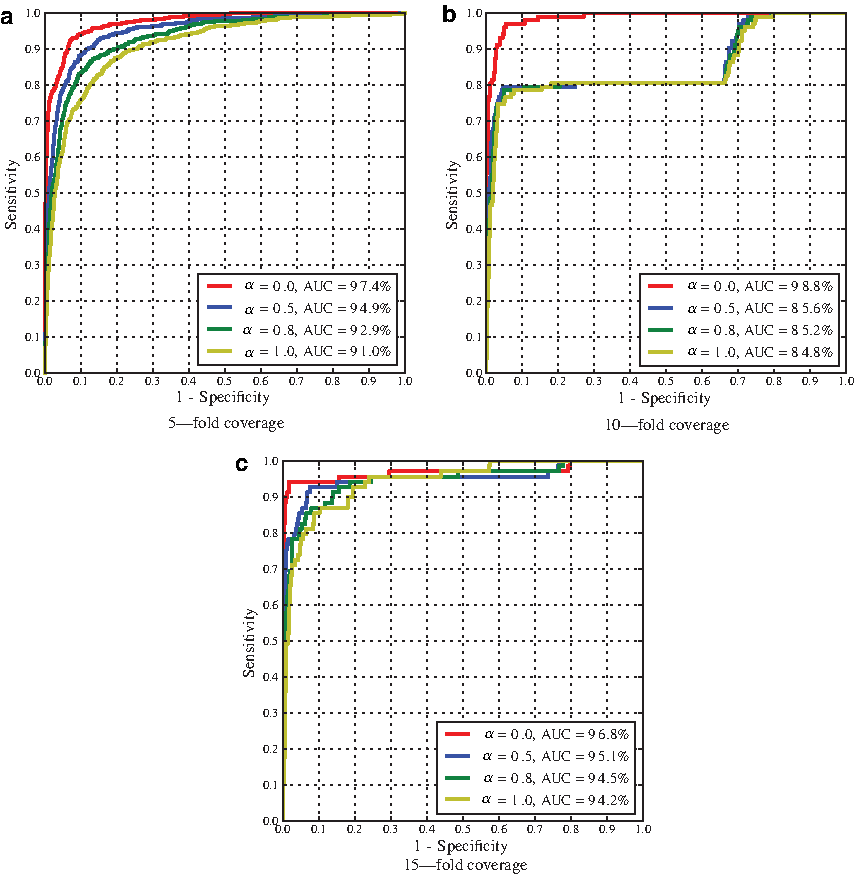
Performance of error-correction restricted to structured positions (i.e., involved in a canonical base pair). Subfigures show accuracy when ART fold parameter is set to 5-, 10-, and 15-fold coverage. We also analyze the impact of the parameter α distributing the weights of stacking pair energies versus isostericity scores and use values ranging from α = {0,0.5,0.8,1.0}. The AUC is indicated in the legend of the figures. Each individual ROC curve represents the average performance over at least 10 experiments.
Our data demonstrates that even on long sequences our algorithm shows interesting potential for error-correction applications. First, the AUC values (up to 0.81) when looking at all positions, in Figure 6, indicate that a signal has been successfully extracted. When restraining to structured regions (i.e., base-paired regions), we obtain AUC values up to 0.988. Since contributions to the energy and isostericity only arise from structured regions, this was an expected result.
An interesting feature is that the best results are almost always obtained when α=0, i.e., when all the contribution comes from the isostericity.
A final observation is related to all positions as in Figure 6. The first 20% to 30% of sensitivities are obtained almost without any errors and correspond to the nucleotides in structured regions. The rest of the predictions are done on unpaired nucleotides.
4. Conclusions
In this article, we presented a new and efficient way of exploring the mutational landscape of RNA under structural constraints and applied our techniques to identify and fix sequencing errors. In addition, we introduced a new scoring scheme to measure the likelihood of sequencing errors that combines the classical nearest-neighbor energy model parameters (Turner and Mathews, 2010) to the recently introduced isostericity matrices (Stombaugh et al., 2009). The latter accounting for geometrical discrepancies occurred during base-pair replacements.
We combined our algorithm for exploring the mutational neighborhood of an input sequence with known secondary structure to this new pseudo energy model and created a tool to predict pointwise sequencing errors in structured RNA. Importantly, our algorithm runs in Θ(n · (|Ω|+M2)) time and Θ(n · (|Ω|+M)) memory, where n is the length of the RNA, M the number of mutations, and Ω the size of the multiple sequence alignment. This achievement enables us to envision applications to high-throughput sequencing pipelines.
We validated our model on the 5s rRNA and 16s rRNA (see Section 3) and showed that our technology enables us to recover mutational errors with high accuracy (area under the ROC curve between 0.95 and 0.99 when restricting predicted mutations to base-paired regions). Interestingly, we observed that using the isostericity matrices alone yields higher performance than with the nearest-neighbor energy model alone. This finding supports our hypothesis that isostericity matrices provide a valuable source of information that can be efficiently used for RNA sequence and structure analysis. Nonetheless, we also found that the nearest-neighbor model seems to provide a valuable signal when the number of errors in the input sequences is underestimated.
Our techniques are designed to correct pointwise errors in structured regions (i.e., base-paired nucleotides). However, our software RNApyro can be easily combined with other methodologies previously developed for correcting other types of sequencing errors such as indels in unstructured regions or repeats (Quinlan et al., 2008; Quince et al., 2009).
In future works, we also intend to include our model errors stemming from insertions or deletions. It is indeed theoretically possible to consider these scenarios within our dynamic programming scheme (Waldispühl et al., 2002), with minor impacts to the complexity. Finally, we hope that integrating our software to current sequencing pipelines used in metagenomics studies will permit us to improve the estimate of microbial diversity.
Footnotes
Acknowledgments
The authors would like to thank Rob Knight for his suggestions and comments. This work was funded by the French Agence Nationale de la Recherche (ANR) through the M
Author Disclosure Statement
The authors declare that no competing financial interests exist.