
Abstract
Abstract
Genome-scale metabolic model reconstruction is a complicated process beginning with (semi-)automatic inference of the reactions participating in the organism's metabolism, followed by many iterations of network analysis and improvement. Despite advances in automatic model inference and analysis tools, reconstruction may still miss some reactions or add erroneous ones. Consequently, a human expert's analysis of the model will continue to play an important role in all the iterations of the reconstruction process. This analysis is hampered by the size of the genome-scale models (typically thousands of reactions), which makes it hard for a human to understand them. To aid human experts in curating and analyzing metabolic models, we have developed a method for knowledge-based generalization that provides a higher-level view of a metabolic model, masking its inessential details while presenting its essential structure. The method groups biochemical species in the model into semantically equivalent classes based on the ChEBI ontology, identifies reactions that become equivalent with respect to the generalized species, and factors those reactions into generalized reactions. Generalization allows curators to quickly identify divergences from the expected structure of the model, such as alternative paths or missing reactions, that are the priority targets for further curation. We have applied our method to genome-scale yeast metabolic models and shown that it improves understanding by helping to identify both specificities and potential errors.
1. Introduction
G
Curation is performed after the automatic inference of the draft metabolic model. Inference methods combine databases of reactions and pathways with genomic information and existing models for similar organisms (Swainston et al., 2011). Genomic data for the new organism is compared to the data of the reference organism, to find genomic evidence such as the presence of catalyzing enzymes for the reactions conserved in the new organism. Starting from the inference of a draft model, the model refinement process includes several iterations of model analysis, error detection, and improvement (Thiele and Palsson, 2010). The models produced at each iteration are intended for computer simulation, and so describe all the reactions thought to participate in the organism's metabolism. Although automatic model inference tools and genomic comparison methods are becoming steadily more sophisticated, they may still leave gaps in the model or add erroneous reactions. Curation by human experts is necessary.
Much of the complexity of the reaction network comes from biochemically similar reactions that operate on slightly different substrates. For example, in the peroxisome compartment of Yarrowia lypolitica model (MODEL1111190000) (Loira et al., 2012) six acetyl-CoA oxidase reactions are present, transforming fatty acyl-CoAs differing in their carbon chain length (decanoyl-CoA, lauroyl-CoA, etc.) into the corresponding unsaturated fatty acyl-CoAs. There are also several similar reactions for other steps of the β-oxidation of fatty acids pathway (Metzler, 2001). Although all of these details are needed for accurate computer simulation and are common to many models, not all of them are interesting for a curator. It is instead the differences from the common pattern that demand attention. They may be errors in the model, such as missing steps or erroneous connections between pathways, or they may be organism-specific differences such as alternative pathways that are biologically interesting.
To aid human understanding of genome-scale models, while keeping the details needed for a computer simulation, we propose a 3-level zoomable approach:
• The most abstract level represents compartmentalization of the model and focuses on such questions as: Are all the compartments present? Are they well connected by transport reactions? • The second level shows the modules inside of each of the compartments. The questions to be addressed on this level include: Are all the essential processes present? Is the structure of each process correct? Is there any organism-specific adaptation of the structure? • The most detailed level is intended for computer simulation and represents the inner structure of each of the modules with all the substrates, reactions and their kinetics, stoichiometry, and constraints.
The two abstract levels are intended for a human expert, and the last one for the computer.
In this study we focus on the second level of abstraction, which represents the modules inside compartments. A fair amount of work has been done on identifying reusable modules. These approaches can be divided into two groups: series and parallel. A series approach operates on chains of reactions, and generalizes them as a series, consequently hiding the structure of the network. An example of a series approach is representing the network as a set of metabolic pathways [KEGG (Kanehisa et al., 2012); MetaCyC (Caspi et al., 2012)], which can be further divided, for example, into reaction modules (conserved sequences of reactions along the metabolic pathways) (Muto et al., 2013).
The other type of approach operates on reactions that are parallel, keeping the steps and preserving the general view of the network. An example of this approach is grouping reactions based on Enzyme Commission (EC) numbers (Tohsato et al., 2000). The drawback of this approach is that it is not applicable to networks with no EC number assigned or reactions with no catalyzing enzymes identified. We have developed another parallel-reaction method for knowledge-based generalization of metabolic models, which does not depend on enzyme information. It provides a higher-level view of a model while keeping its essential structure and omitting the details.
Definition 1
The model generalization process groups chemical species present in the model into equivalence classes and merges each class into a generalized chemical species. Reactions that involve the same generalized chemical species are then factored together into a generalized reaction.
By applying the model generalization process, we can build a simplified model that focuses on the high-level relationships. The simplified model can be further divided into pathways.
2. Mathematical Basis
2.1. Basic definitions
We represent a metabolic model M as a pair of two sets: a set S of biochemical species, and a set R of reactions between them:
We represent each reaction
To perform the model generalization, we define an equivalence operation ∼ on the species set, and group species into equivalence classes:
Species equivalence imposes reaction equivalence: two reactions are equivalent if their corresponding reactant and product species sets are pairwise equivalent.
Equivalent reactions are factored together into a generalized reaction that operates with generalized species (i.e., species equivalence classes):
In order to maintain the number of distinct species participating in a reaction, restriction (1′), analogous to restriction (1), must be satisfied:
In order to avoid creation of paths in the generalized model that are not based on the evidence from the initial model, we introduce restriction (2): Species that do not participate in any pair of equivalent reactions and do not have any common equivalent species must not be grouped together.
Note that restriction (2) can be reformulated as maximizing the number of species equivalence classes while keeping the reaction equivalence classes unchanged.
The generalized model M/∼ is a pair of generalized species and reaction sets (quotient sets):
The generalized model is a
Every reaction of the generalized model corresponds to at least one reaction of the initial model, having the same topology (number of distinct reactant and product species) and operating on species that can be zoomed out into those participating in the generalized reaction. The appropriate level of abstraction is defined with respect to the initial model as the most general one that satisfies restrictions (1′) and (2).
The method and restrictions are described in Figure 1.
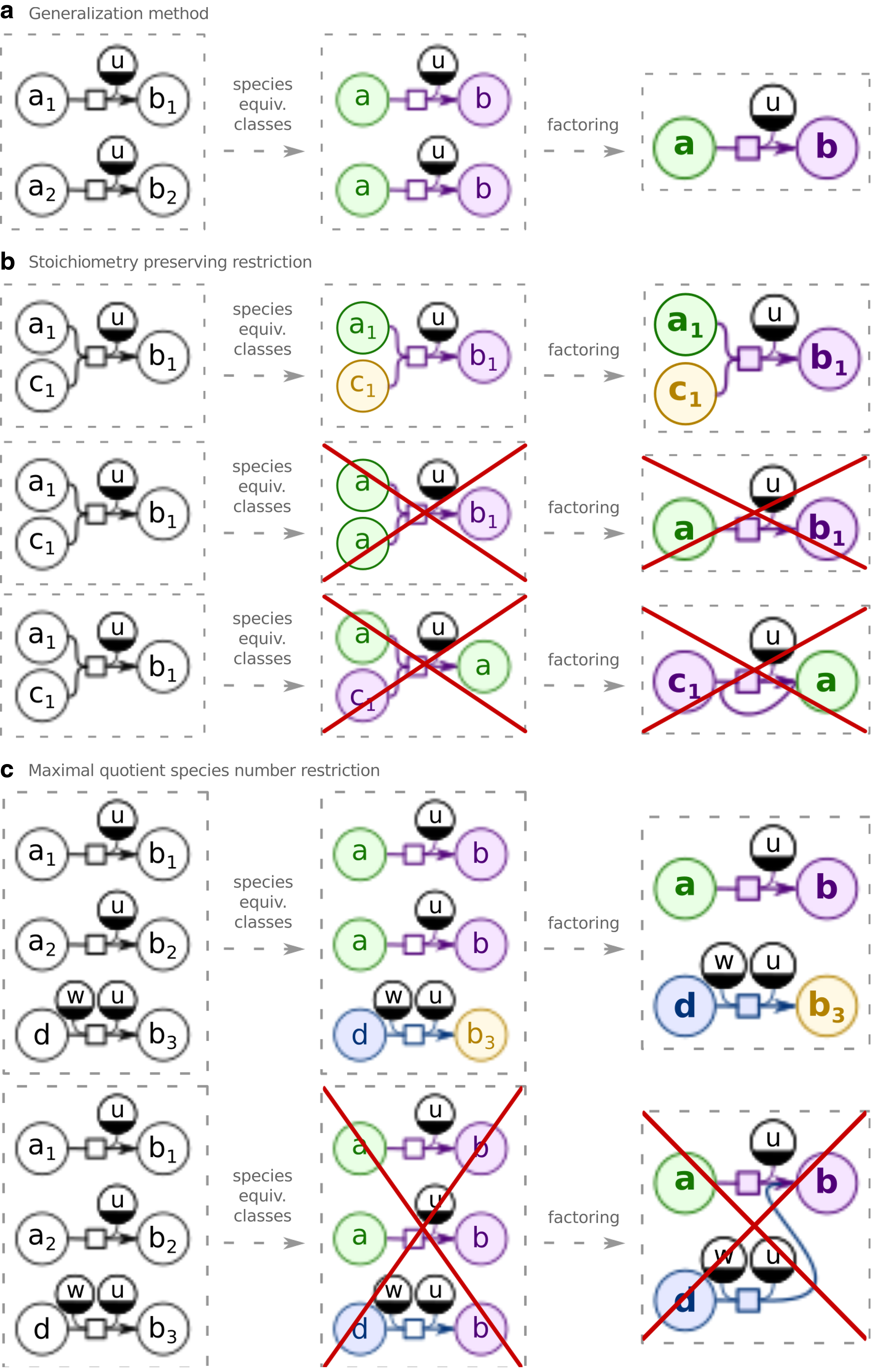
Model generalization method and restrictions.
2.1.1. Specific and ubiquitous species
We say that a ubiquitous species is one that participates in many reactions (more than some threshold), such as water, hydrogen, oxygen, etc. Grouping such species would increase the number of reactions each group would participate in, beyond the sharing already common to most of the models, and decrease readability; in fact, during visualization these species are often even duplicated to improve readability (Rohn et al., 2012). Consequently we do not generalize ubiquitous species. In the generalized model each forms a trivial equivalence class:
Specific species are the others, which we divide into nontrivial equivalence classes and generalize accordingly.
2.2. Model generalization problem
Problem 1
Given a metabolic model M = 〈S,S(ub) ⊂ S,R〉 that describes n species (including
We will solve model generalization problem 1 in three steps:
1. Define the most general equivalence operation 2. Modify the current equivalence operation to satisfy restriction (1′); 3. Modify the current equivalence operation to satisfy restriction (2).
2.2.1. Step 1. Equivalence operation
\documentclass{aastex}\usepackage{amsbsy}\usepackage{amsfonts}\usepackage{amssymb}\usepackage{bm}\usepackage{mathrsfs}\usepackage{pifont}\usepackage{stmaryrd}\usepackage{textcomp}\usepackage{portland, xspace}\usepackage{amsmath, amsxtra}\pagestyle{empty}\DeclareMathSizes{10}{9}{7}{6}\begin{document}
$$\mathop\sim^{\circ}$$
\end{document}
Definition 2
Given a model
Lemma 1
For any equivalence operation ∼ on the model M = 〈S, S(ub) ⊂ S, R〉, the corresponding quotient species set S/∼ and quotient reaction set R/∼ are partitions of, respectively, the quotient species set

The Compute
2.2.2. Step 2. Stoichiometry-Preserving Property Obedience
Problem 2
Given an equivalence operation ∼ defined on a metabolic model M = 〈S,S(ub) ⊂ S,R〉, find an equivalence operation
2.2.2.1. Algorithm
We start with the given equivalence operation ∼0 = ∼, and iteratively improve it until the stoichiometry-preserving property (1′) is obeyed. We denote the equivalence operation obtained at the i-th iteration step as ∼ i .
At each iteration, if there exists a species equivalence class that violates the stoichiometry-preserving property (1′), that is:
we partition this species equivalence class
At each iteration one equivalence species class is partitioned. In the worst case, the equality operation = (each species is equivalent only to itself) will be achieved. As it obeys restriction (1′), the process will terminate.

2.2.2.2. Species equivalence class partition
In a species equivalence class that violates the restriction (1′) there are usually only a few conflicts present, and multiple solutions of the partition problem exist.
2.2.2.2.1. Species ontology
In order to make the choice of the species equivalence classes biologically meaningful, we use an ontology that describes hierarchical is_a relationships (more specific to more general) between biochemical species.
Definition 3
A term t is a model term if it corresponds to a specific species in the metabolic model.
We assume that no two model terms are connected by a descendant–ancestor (more specific–more general) relationship in the ontology; otherwise, we mark the ancestor term ubiquitous:
We iteratively remove all the leaf terms that are not model terms from the ontology so that all the model terms become leaves, and all the leaves become model terms.
For each species equivalence class that needs to be partitioned, we first find the least common ancestor T of the ontological terms corresponding to its species. If the ontology allows for multiple inheritance, and there are several such least common ancestors, we pick the first one. Then we look among the T-th descendant terms for those that are compatible (to avoid multiple inheritance).
Definition 4
Terms
Problem 3
Given a term T, find a compatible term set among its descendants, such that it has minimal size, covers all the T-th descendant leaf terms, and satisfies the stoichiometry-preserving property (1″):
To do so, we first exclude all the terms that violate the stoichiometry-preserving property (1″). We thus obtain an exact set cover problem.
Problem 4 (Set cover)
Given a set X and a collection of its finite subsets Ψ, such that ∪S∈Ψ S = X, find a minimum-size subset Π ⊂ Ψ whose members cover all of X: ∪S∈Π S = ∪S∈Ψ S = X.
Remark 1
Set cover is NP-complete (Karp, 1972).
Problem 5 (Exact set cover)
As in Set cover problem, except that here the sets used in the cover are not allowed to intersect.
Remark 2
Exact cover is NP-complete (Goldreich, 2008).
2.2.2.2. Exact set cover applied to ontological terms
Each ontological term t defines a set S(t) of its descendant leaf terms (including t if it is a leaf). The instance consists of a set X of the model terms of interest, and a collection Ψ of all sets defined by their common ancestor T, its descendant terms, and their relative complements with respect to
Note that in this case an exact cover always exists, for example, the one formed by all the leaf terms.
2.2.2.3. Choice of the ontology
We assume that any term that violates property (1″) is removed from the ontology. Note that the term T is also removed.
If the ontology has no multiple inheritance, that is,
We use the ChEBI ontology (de Matos et al., 2010) of chemical compounds, as it is a de facto standard for species annotation in metabolic models. ChEBI consists of three main branches: chemical entity, role, and subatomic particle. The chemical entity branch describes terms useful for annotation of biochemical species in a metabolic model.
The level of detail in the ChEBI hierarchy is not uniform: some sub-branches are more developed than others, so equally precise terms may be placed unequally deep in the hierarchical tree. For example, both hydrogen peroxide (CHEBI:16240) and decanoyl-CoA (CHEBI:28493) terms describe precise chemical molecules, but hydrogen peroxide is only 5 terms away from the chemical entity in the ChEBI hierarchy, while decanoyl-CoA is 11 terms away.
Besides that, different types of classification are combined together in the hierarchical tree, leading to multiple inheritance. For example, in the fatty-acid (CHEBI:35366) sub-branch, several classification types are present, including:
• classification based on the length of the carbon chain: – short-chain fatty acid (CHEBI:26666): 2–4 carbons; – medium-chain fatty acid (CHEBI:59554): 6–12 carbons; – etc. • classification based on the presence of double bonds in the carbon chain: – saturated fatty acid (CHEBI:26607): no double bonds; – unsaturated fatty acid (CHEBI:27208): one or more double bonds; • classification based on substituent groups: – hydroxy fatty acid (CHEBI:24654): one or more hydroxy substituents; – oxo fatty acid (CHEBI:59644): at least one aldehydic or ketonic group; – etc.
Moreover, using only hierarchical relationships in the ChEBI ontology is not always enough. Examples show that similar reactions can happen to the acid and the base in a conjugate acid–base pair. A conjugate acid–base pair is two species, one an acid and one a base, that differ from each other through the loss or gain of a proton (Stoker, 2012). For instance, in the Rhea database of chemical reactions (Alcántara et al., 2012), the acyl-CoA oxidase (RHEA:28354) reaction: decanoyl-CoA + FAD + H + → trans-dec-2-enoyl-CoA + FADH2 is found for both decanoyl-CoA (CHEBI:28493) and its conjugate base decanoyl-CoA(4-) (CHEBI:61430). But hierarchically these species are very far from each other in the ChEBI ontology. Their least common ancestor is molecular entity (CHEBI:23367), a direct descendant of the root chemical entity. To establish a conjugate acid–base pair correspondence in the ChEBI ontology, not the hierarchical (is_a) but the special is_conjugate_base_of /is_conjugate_acid_of relationships are used. To maximize the chances of a conjugate acid–base pair being in the same quotient species set, we generalize the hierarchical relationship.
Definition 5
Term t is a generalized direct descendant/ancestor of a term T if and only if t or a conjugate base or acid of t is a direct descendant/ancestor of T or of a conjugate base or acid of T.
Definition 6
Term t is a generalized descendant/ancestor of a term T if and only if t is a generalized direct descendant/ancestor of T or of any generalized descendant/ancestor of T.
We extend Ψ so that it is closed under the operation of relative complement:
2.2.2.4. Greedy algorithm
Among the available subset candidates

Greedy set cover is a polynomial time approximation algorithm that achieves an approximation ratio of
2.2.3. Step 3. Species Equivalence Class Number Maximization
Problem 6
Given an equivalence operation ∼ defined on a metabolic model
2.2.3.1. Algorithm
To satisfy restriction (2) we associate each species s in the initial model with a pair of sets of reaction equivalence classes in the quotient reaction set R/∼, induced by reactions in which it participates as a reactant or product.
Definition 7
Given an equivalence operation ∼ defined on a metabolic model
Any further partition of the quotient species set would imply the partition of the quotient reaction set. Hence the number of species equivalence classes is maximal for the current number of reaction equivalence classes, and restriction (2) is satisfied.

2.2.4. Complete algorithm
3. Applications
To illustrate the model generalization method we show its application to the genome-scale metabolic network of the lipid-accumulating yeast Yarrowia lipolytica [MODEL1111190000 (Loira et al., 2012)]. The generalized model is included as Supplementary Material (available online at www.liebertpub.com/cmb). The generalization has created 100 nontrivial quotient species and 217 nontrivial quotient reactions, and reduced the total number of species from 1847 to 1072 and of reactions from 2002 to 893.
The generalization method shows the best performance if the model is well annotated with ChEBI. The species lacking ChEBI annotations are forced to form trivial quotient species in the generalized model as there is no evidence of their biochemical similarity with any other species in ChEBI. For 430 species in the Y. lipolytica model, no appropriate ChEBI annotation was found, thus they could not be grouped with other species.
Peroxisome is an example of a well-annotated compartment in the Y. lipolytica model. Only two species have no ChEBI annotations: YLR043C disulphide and TRX1. The generalization process reduced the number of reactions in peroxisome from 65 to 27. Figures 2 and 3 represent the peroxisome before and after the generalization and were produced using Tulip graph visualization tool (Auber, 2004).
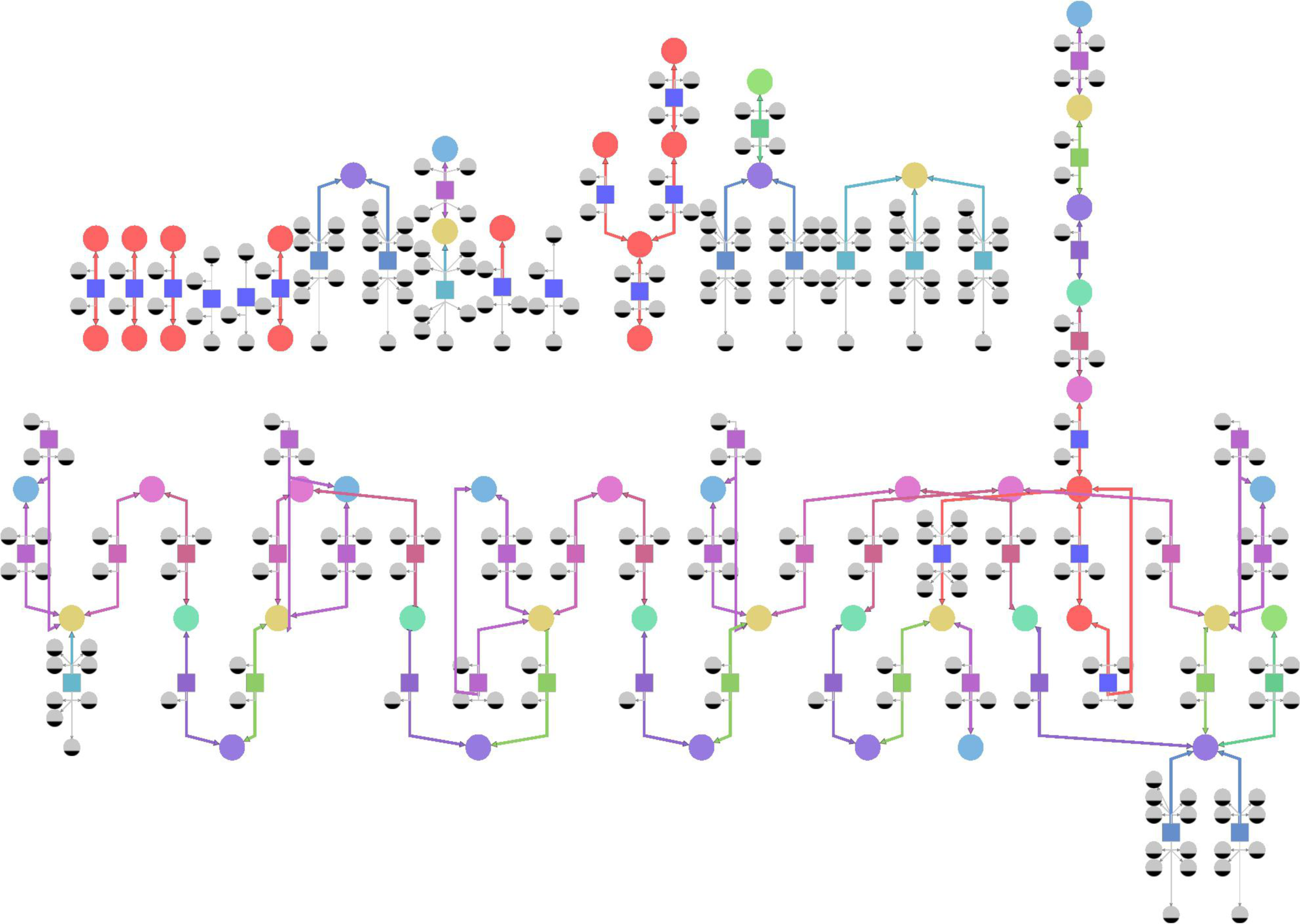
Peroxisome of the Y. lypolitica model (MODEL1111190000). Species are represented as circular nodes, and the reactions as square ones, connected by edges to their reactants/products, according to Systems Biology Graphical Notation (SBGN) notation (Moodie et al., 2011). Ubiquitous species are of smaller size and gray in color. Specific species are divided into six nontrivial equivalence classes, and colored accordingly (violet, light-blue, yellow, green, light-green, magenta). The specific species that form trivial equivalence classes are all colored red. Reactions are divided into 15 nontrivial equivalence classes, also represented by different colors. Reactions that form trivial equivalence classes are all colored blue. The size of the model does not allow for readable species labels, so they are omitted.
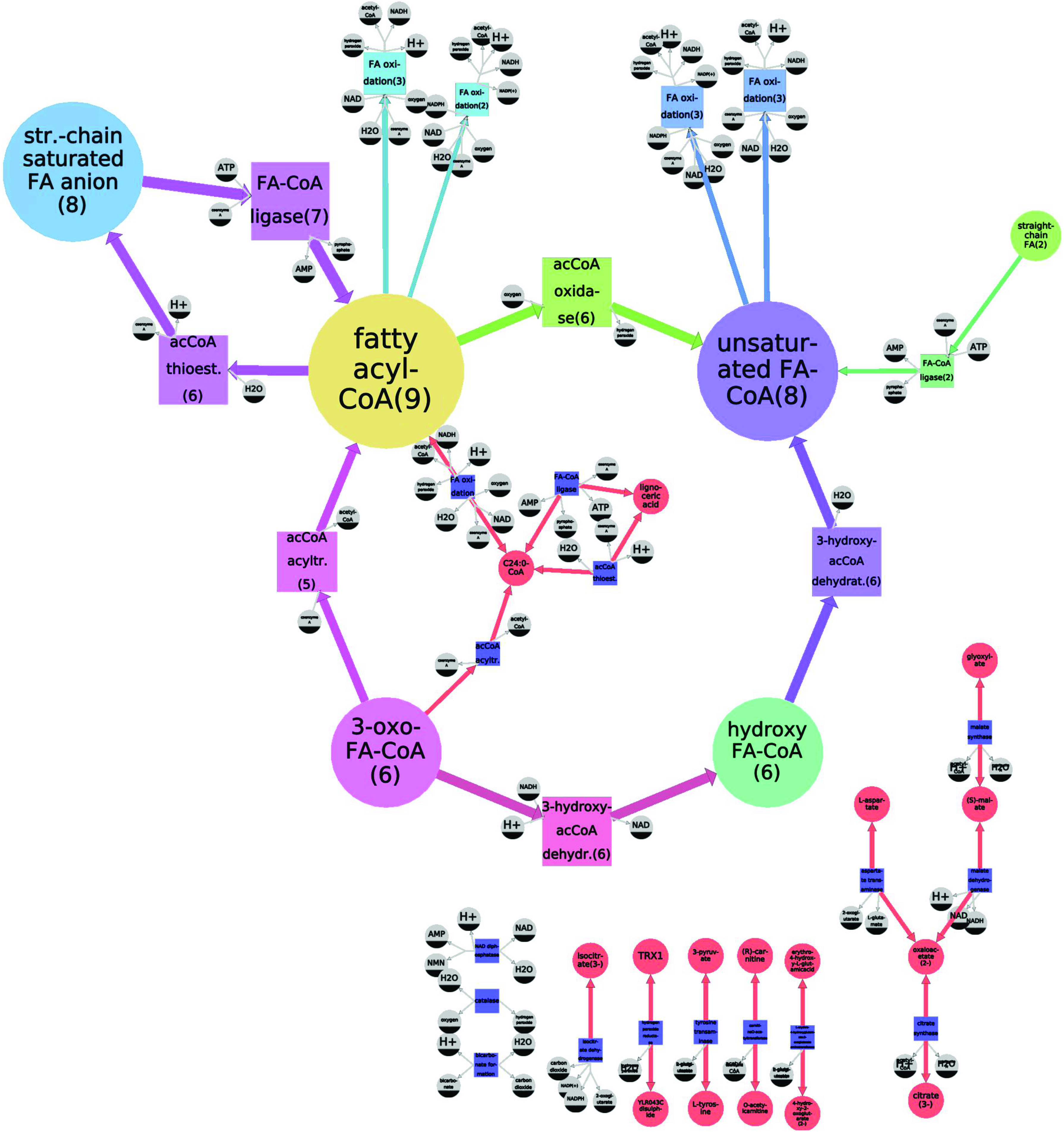
The generalization of the peroxisome of the Y. lypolitica model (see Fig. 2). The generalized model operates on quotient species and reactions. The number given in parentheses and the size of each node indicates how many entities it generalizes.
The model before the grouping of equivalent reactions and species into generalized ones is shown on Figure 2: different colors correspond to different equivalence classes. The same color code is used in Figure 3 representing the generalized model that operates with quotient species and reactions. For example, the violet unsaturated FA-CoA node is a quotient of 8 species: hexadec-2-enoyl-CoA, oleoyl-CoA, tetradecenoyl-CoA, trans-dec-2-enoyl-CoA, trans-dodec-2-enoyl-CoA, trans-hexacos-2-enoyl-CoA, trans-octadec-2-enoyl-CoA, and trans-tetradec-2-enoyl-CoA (colored violet in Figure 2). In a similar manner, the light-green acCoA oxidase quotient reaction, which converts fatty acyl-CoA (yellow) into unsaturated FA-CoA (violet), generalizes six corresponding light-green reactions of the initial model (Figure 2).
The generalized model describes the β-oxidation of fatty acids pathway (Metzler, 2001) happening inside the Y. lypolitica peroxisome in a generic way: as a transformation of fatty acyl-CoA (yellow) into unsaturated FA-CoA (violet), then into hydroxy FA-CoA (green), 3-oxo FA-CoA (magenta), and back to fatty acyl-CoA (with a shorter carbon chain); while the initial model describes the same process in more details, specifying those reactions for each of the fatty acyl-CoA species present in the organisms' cell (e.g., decanoyl-CoA, dodecanoyl-CoA, etc.). That is why the beta-oxidation chain of the reactions in the initial model, transforming step-by-step the fatty-acyl-CoA with the longest carbon chain into the one with the shortest chain, in the generalized model appears as a cycle (generalizing all the fatty-acyl-CoAs into one species, regardless the chain length).
The more precise model is needed for simulation, while the more general one is clearer to a human and reveals the main properties of the model. For example, the generalized model highlights the fact that there is a particularity concerning C24:0-CoA (tetracosanoyl-CoA) (red, inside the cycle in Figure 3): there exists a “shortcut” reaction, producing it directly from another fatty acyl-CoA (yellow), avoiding the usual four-reaction beta-oxidation chain used for other fatty acyl-CoAs.
Another application of model generalization is metabolic model comparison. The generalization brings the models to the same level of abstraction and highlights the differences such as gaps. Examples can be found in (Zhukova and Sherman, 2013).
6. Discussion
We have developed a method that provides a semantically zoomed-out view of a metabolic model, that keeps its essential structure but hides the details.
We have implemented our method as a Python program, which is available for download online from http://metamogen.gforge.inria.fr. It takes a model in SBML format (Hucka, 2003) as an input, annotates its species with ChEBI terms (if the annotations are not present in the model), and generalizes it. It produces two SBML files as an output. The first output file contains the generalized model. The second output file uses groups (Hucka, 2012) extension of SBML and contains the initial model plus the groups representing quotient species and reaction sets.
We have applied our method to genome-scale metabolic models of yeasts. We have illustrated it here on the lipid-accumulating yeast Y. lipolytica and have shown that generalization helps to find gaps and peculiarities, as well as compresses the information stored in the model, which can be used for model visualization and model comparison. In the example, the chain of β-oxidation of fatty acids reactions in the constitutive peroxisome of Y. lipolytica is generalized to a cycle of reactions, highlighting the alternative path for C24:0-CoA (tetracosanoyl-CoA).
Currently the generalization method depends on the ChEBI ontology. It cannot generalize species that lack ChEBI annotations. In future work we will overcome this limitation.
The method zooms out a model to the most general level of abstraction that coexists with the model structure, that is, does not violate the restrictions (1′) and (2). It remains to be seen whether there are intermediate levels of abstraction that can be useful for model analysis. In particular it may be interesting to define the maximal generalization for a group of organisms to highlight the specific differences of the individual models with respect to a common generalization.
Footnotes
Acknowledgments
The authors would like to thank Drs. Romain Bourqui and Antoine Lambert of the LaBRI MABioVis team for advice on graph layout. A.Z. was supported by a CORDI-S doctoral fellowship from Inria.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.