
Abstract
Abstract
Single-nucleotide polymorphisms (SNPs) are implicated in the complexity of understanding the genetics of diseases and their therapeutics. Here we have attempted to determine the impact of nonsynonymous SNPs (nsSNPs) on structure, dynamics, and ligand-binding properties of the human acetylcholinesterase (hAChE) protein, which has been targeted in the treatment of Alzheimer's disease. Of the reported 153 SNPs, 4 nsSNPs, namely, A415G, P104A, V302E, and Y119H, were prioritized to be functionally unfavorable by SIFT and PolyPhen algorithms. Molecular dynamics simulation revealed these nsSNP forms to be structurally stable, and they are also considered functionally active as they lie away from the catalytic triad. However, the aromatic amino acids lining the active-site gorge exhibited altered degrees of side chain dihedral angles. Such conformational alterations were evaluated for their ability to interfere with binding of hAChE inhibitors. The inhibitors (donepezil, galantamine, rivastigmine, and tacrine) were oriented differently in comparison to the native because of the steric hindrance offered by the altered dihedral angles. Interestingly, huperzine A alone exhibited higher efficiency in its binding to the AChE and retained similar orientation irrespective of the polymorphisms since the orientation of Asp74 involved in its binding and trafficking remained unaltered in all protein forms. Therefore, we conclude that nsSNPs confer changes to the dynamicity of proteins, which in turn affects their ligand-binding properties rather than their stability. Considering the diverse polymorphic nature of hAChE, while contemplating any structure-based drug design, the common, nonpathogenic nsSNPs should be considered for the utmost efficacy of drugs.
1. Introduction
S
The AChE (EC 3.1.1.7) protein, coded by the ACHE gene located at 7q22, after expression, is localized at high concentrations in cholinergic neurons (Getman et al., 1992). In addition, it is also found at low and variable concentrations in adrenergic and sensory neurons, respectively (Soreq et al., 1990). AChE belongs to the class of serine hydrolases and plays a fundamental role in synaptic transmission by way of hydrolyzing its substrate, a neurotransmitter, acetylcholine. It is one of the fastest enzymes known, having a high turnover number. This reflects its catalytic ability of hydrolyzing an acetylcholine molecule in 80 ms by an unusual catalytic triad—Ser, His, and Glu, instead of Asp. This catalytic triad is located at a 20-Å-deep gorge below the surface of the protein that comprises of 614 amino acids (Quinn, 1987). A unique surface region of the protein called the peripheral anionic site (PAS) (Taylor and Lappi, 1975) leads to the catalytic triad through a gorge lined by 14 aromatic amino acids that are unique for this protein (Sussman et al., 1991).
Classical hAChE inhibitors (hAChEI) are regularly prescribed for the symptomatic treatment of AD, although several other proteins, such as NMDA receptor, β secretase, and amyloid beta peptides, are predicted to be potential targets in AD. The FDA-approved drugs, namely, donepezil, galantamine, rivastigmine, and tacrine, which are dominating the market today, all belong to hAChEI. Huperzine A, an alkaloid drug traditionally used in Chinese medicine against cholinesterases (Wang et al., 1986), is currently in clinical trials for its future use in AD treatment (Rafii et al., 2011). Presently, tacrine is not frequently prescribed as it has serious side effects. Donepezil, galantamine, and rivastigmine were also shown to have side effects, which include nausea, vomiting, and loss of appetite, but still these are widely accepted as drug of choice. However, the efficacy of any such drugs should be evaluated case by case in individuals for better symptomatic improvement as their effects are not identical in all patients (O'Donovan, 2011), which urges the need for the present study.
The differential effect of these drugs on AD patients shall be attributed to the polymorphic nature of hAChE. The SNPs in hAChE that are inherited in AD individuals may possibly cause conformational changes in the protein that might alter its interaction with AD drugs. As such hAChE has several nsSNPs whose pathogenicity with relevance to AD has not yet been attempted upon. In the present study, an attempt has been made toward identification of unfavorable nsSNPs of the hAChE protein and such nsSNPs are evaluated for their effect on stability, dynamics, and interaction of the protein with best-known AD drugs, toward understanding their efficacy as the best drug.
2. Methods
2.1. SNP datasets
The SNPs associated with the human ACHE gene were collected online from the SNP database, dbSNP (Sherry et al., 2001).
2.2. Analysis of functional consequences of nsSNPs using SIFT and PolyPhen
SIFT (Ng and Henikoff, 2003) and PolyPhen algorithm (Ramensky et al., 2002) were used for predicting functionally unfavorable nsSNPs based on sequence and structure homology. All the nsSNPs were analyzed, predicted, and prioritized by SIFT based on the tolerance index of ≤0.05, which was considered to be functionally deleterious. Similarly, using PolyPhen tool, an undesirable nsSNP had a position-specific independent count score difference (PSIC SD) of ≥0.5.
2.3. Sequence conservation analysis
Multiple sequence alignment (MSA) and conservation analysis was done by ClustalX v2.0.11 (Larkin et al., 2007). The study included representative sequences from different species. Their UniProt IDs are given in parentheses: torpedo (P04058), marbled electric ray (P07692), electric eel (O42275), human (P22303), bovine (P23795), mouse (P21836), rat (P37136), chick (P36196), and house fly (P07140).
2.4. Homology modeling
The Cartesian coordinates for the hAChE protein in complex with fasciculin II were obtained from Protein Data Bank (PDB ID: 1B41) and used as a template for modeling all the mutant proteins corresponding to different nsSNP forms. Using Modeller9v9 (Eswar et al., 2007), 10 models were built by homology modeling. Such models were generated by filling the missing residues in the crystal structure and also by replacing the appropriate amino acids corresponding to nsSNP forms. While modeling, the two water molecules that have a high level of conservation in all the vertebrates that are capable of forming hydrogen-bonding network with the amino acids Glu202, Glu450, and Tyr133 were retained in the structure (Kryger et al., 2000). Then, the models were stereochemically validated by PROCHECK analysis (Laskowski et al., 1993).
2.5. Molecular dynamics simulation
The models thus generated were the starting structures for molecular dynamics (MD) simulation carried out by GROMACS package (Van Der Spoel et al., 2005) using GROMOS96 53a6 force field (Lindahl et al., 2001). In brief, the native and mutant hAChE proteins were solvated with Simple Point Charge water model (Berendsen et al., 1981) within a dodecahedron box such that there exists a distance of 1 nm between the protein and the wall. The overall charge of the system was neutralized by adding counter ions. The whole system was then relaxed for ensuring proper geometry through energy minimization by steepest descent with a tolerance of 1000 kJ/nm. The minimized system was then equilibrated for a period of 200 ps at 300 K temperature and 1 bar pressure by position-restrained MD simulation for favorable positioning of the solvent and ions about the solute. The equilibrated system was then subjected to MD simulation for a period of 10 ns. The temperature and pressure of the system was maintained constant by coupling the protein and nonprotein component of the system separately to v-rescale thermostat (τ = 0.1 ps) and to Parrinello–Rahman algorithm (τ = 2.0 ps) with a compressibility of 4.5 × 10−5 bar −1. Neighbor list was determined by grid method with a cutoff range of 1 nm for short-range interactions. Long-range electrostatic interactions were handled by Particle Mesh Ewald algorithm (Darden et al., 1993; Essmann et al., 1995), and the boundary effect was eliminated by periodic boundary condition. The integration time was 2 fs for all calculations, and the coordinates were saved every 2 ps during the MD simulation. The LINCS method (Hess et al., 1997) was used for fixing all the covalent bonds.
2.6. Molecular docking
LigPrep (v2.3) and Glide (v5.5) modules of Schrodinger (Schrödinger, Inc.) were used for all the docking calculations. Before docking, the proteins were subjected to optimization and refinement using “Protein Preparation” wizard. The structures were then subjected to root mean square deviation (RMSD)-restrained minimization using Impref, and the final structures thus obtained formed the basis for the docking studies. All possible low-energy conformations of ligands were prepared using LigPrep. A flexible, extraprecision docking (Friesner et al., 2006) was performed using Glide for obtaining the GlideScore of different mutant proteins with five different AD drugs. Lowest score was set as the criteria to predict the favored binding modes of each drug. The docking score in terms of GlideScore was used to rank the docked poses. The top-ranking conformants with lowest binding energy were visually analyzed with Glide extraprecision visualizer, and the best-possible pose for each protein–ligand complex was thus obtained.
3. Results and Discussion
3.1. Distribution of SNPs in the ACHE gene
A total of 153 SNPs in the human ACHE gene reported in dbSNP were retrieved. Of the total 153 SNPs, 9 were silent (i.e., synonymous SNPs, 5.89%) and 17 were missense (i.e., nsSNPs, 11.11%), both in the coding region. Introns accounted for 70 SNPs (45.75%), and 54 (35.29%) were in the other noncoding regions (5′ and 3′ UTRs). The rest (1.96%) accounted for 1 frame-shift and 2 stop-gain mutations. Among the nsSNPs, only 15 that can be mapped into the crystal structure of the hAChE protein were considered in the present study for further computational analysis. Other 2 nsSNPs were excluded since they lie at the extreme N-terminal region that is not forming any part of the crystal structure available.
3.2. Functional integrity of nsSNPs of hAChE as predicted by SIFT and PolyPhen
Since these nsSNPs of hAChE were not implicated in any pathogenesis, the nature and functional consequences of these nsSNPs were probed using SIFT and PolyPhen toward prediction of associated ill-effect. SIFT identified only 6 nsSNPs out of 15 to be detrimental to the functional integrity of hAChE with a tolerance index ranging from 0 to 0.04 (Table 1). Among them, mutants A415G and V302E (numbering refers to the hAChE crystal structure, 1B41, and the same is used throughout the article) were highly detrimental with a score of 0. PolyPhen algorithm identified 5 out of 15 nsSNPs as unfavorable. According to this tool, the mutants such as V302E, P104A, Y119H, A415G, and G89E were predicted to be unfavorable based on their PSIC SD of 1, 0.997, 0.997, 0.962, and 0.56, respectively (Table 1). Thus, both SIFT and PolyPhen together identified only 4 (A415G, P104A, V302E, and Y119H) out of 15 nsSNPs that could have an adverse impact on the hAChE protein and were considered further. All other mutants were predicted to have negligible effect in influencing the function of hAChE and hence were not considered for further analysis.
The unfavorable nsSNPs predicted by SIFT and/or PolyPhen are in bold. AA change, amino acid change; nsSNPs, nonpathogenic nonsynonymous single-nucleotide polymorphisms; PSIC SD, position-specific independent count score difference.
3.3. Characterization of amino acid residues in native and nsSNP forms of hAChE
MSA analysis of the AChE protein among different species revealed that all four nsSNPs were distributed among regions of conserved sequences. However, the residues A415 and V302 were occasionally replaced by other residues (Fig. 1). Strict conservation was observed among residues at the 119th position, whereas the residue at the 104th position holds no conservation across the species. In A415G, the amino acid glycine being smaller than alanine may offer more flexibility to the mutant protein. However, in P104A, no change in polarity but in hydrophobicity is observed. Interestingly, these two amino acids (G and A) in A415G and P104A are more favorable for α-helix formation than the native amino acids. In V302E, the negatively charged glutamic acid residue replaces valine, a hydrophobic amino acid, and hence this nsSNP could disrupt the hydrophobicity of the protein. Similarly in Y119H, although the polarity is maintained, the hydrophobicity is disturbed by the basic amino acid histidine, which is an excellent nucleophile. In spite of such theoretical predictions, the exact impact of these nsSNPs on the structure needs further evaluation.

Multiple sequence alignment showing level of conservation of AChE amino acid sequences. The polymorphisms under investigation are indicated by red arrows. The notations asterisk (*), colon (:), and period (.), respectively, indicate strictly, highly, and moderately conserved amino acid residues. The amino acids are colored according to their physiochemical properties as defined by the ClustalX color scheme.
Virtual inspection of the protein structure indicates that all these nsSNPs are distributed only on the outer surface of the hAChE protein (Fig. 2) and no mutations (except Y119H) occur at close proximity (i.e., within 5Å) to the catalytic triad. Though these mutated amino acids are distributed at conserved regions, as they occur on the outer surface of the protein, they may not directly interfere with the catalytic activity. Also, all the native amino acids are nonreactive and so do not participate directly in the functioning of the protein. Therefore, any replacement in them might not affect the protein function. These predictions are partially true and are in agreement with earlier report that the well-known nsSNP corresponding to H353N does not affect the catalytic efficiency of the hAChE protein (Masson et al., 1994). Nevertheless, all the four nsSNPs that are the focus of the present study cannot be ruled out for possible additional roles, namely, the differential inhibition of hAChE by AD drugs.
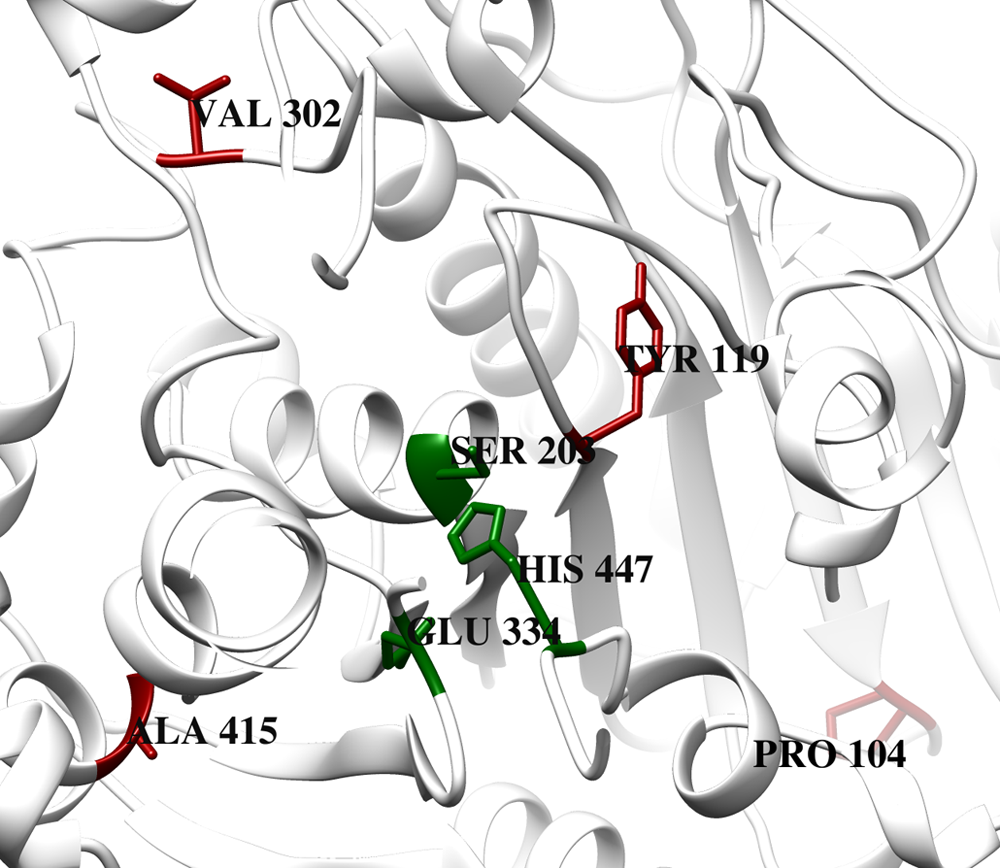
The location of nsSNPs on the protein structure. The corresponding positions of four different nsSNPs are shown in red and the active-site residues are represented in green in a native hAChE structure. All of the mutated residues occupy the outer surface area of the protein. nsSNPs, nonpathogenic nonsynonymous single-nucleotide polymorphisms.
3.4. Homology modeling of nsSNP forms of hAChE
Mutations were manually inserted and appropriate mutant models were constructed and selected based on the most favored stereochemical arrangement of the atoms. All the modeled structures had a majority of residues (99%) encompassed in the allowed regions of the Ramachandran plot with 90% of the residues in the most favored region. The backbone RMSD values of the mutants A415G, P104A, V302E, and Y119H with respect to native structure were 0.171 Å, 0.195 Å, 0.199 Å, and 0.214 Å, respectively. As the deviation was only about 0.1–0.2 Å from the native protein structure, these mutations were presumed that they do not affect the structure of the proteins. However, their structural stability and the quantum of structural deviations were further analyzed through MD simulation.
3.5. Structural properties of nsSNP forms of hAChE
As most mutations are frequently confined to structural instability, MD simulation was performed with modeled proteins to gain insights into the quantum of structural alterations that had occurred because of the mutation. Structural integrity of the protein was examined based on the RMSD of backbone atoms (Fig. 3a) and radius of gyration (Rg) as a function of simulation time (Fig. 3b). These results demonstrate that these mutations affect the structural integrity of hAChE to a very negligible extent. Among the four mutants, Y119H, V302E, and P104A showed a very slight increase (∼0.05 nm) in the backbone RMSD compared with the native, thus reflecting the overall structural integrity of the protein. However, A415G showed a significantly lower RMSD, implying that this mutation enhances the compactness of the protein during the course of molecular simulation. Since the deviation lies within the tolerable range (<0.05 nm), it is concluded that all these nsSNPs do not affect the stability of the protein. Figure 3b shows similar Rg with no deviations for the two mutants, A415G and V302E when compared to native, while in P104A insignificant deviations were observed. On the other hand, Y119H had comparatively lesser Rg. The difference from the native was only ∼0.03 nm, which might be because of the tendency of histidine to form ionic interactions. The Rg of the protein also reflected the preserved integrity of the mutant proteins, which further substantiates the results of Figure 3a. These minor deviations may not disturb the stability of the protein.
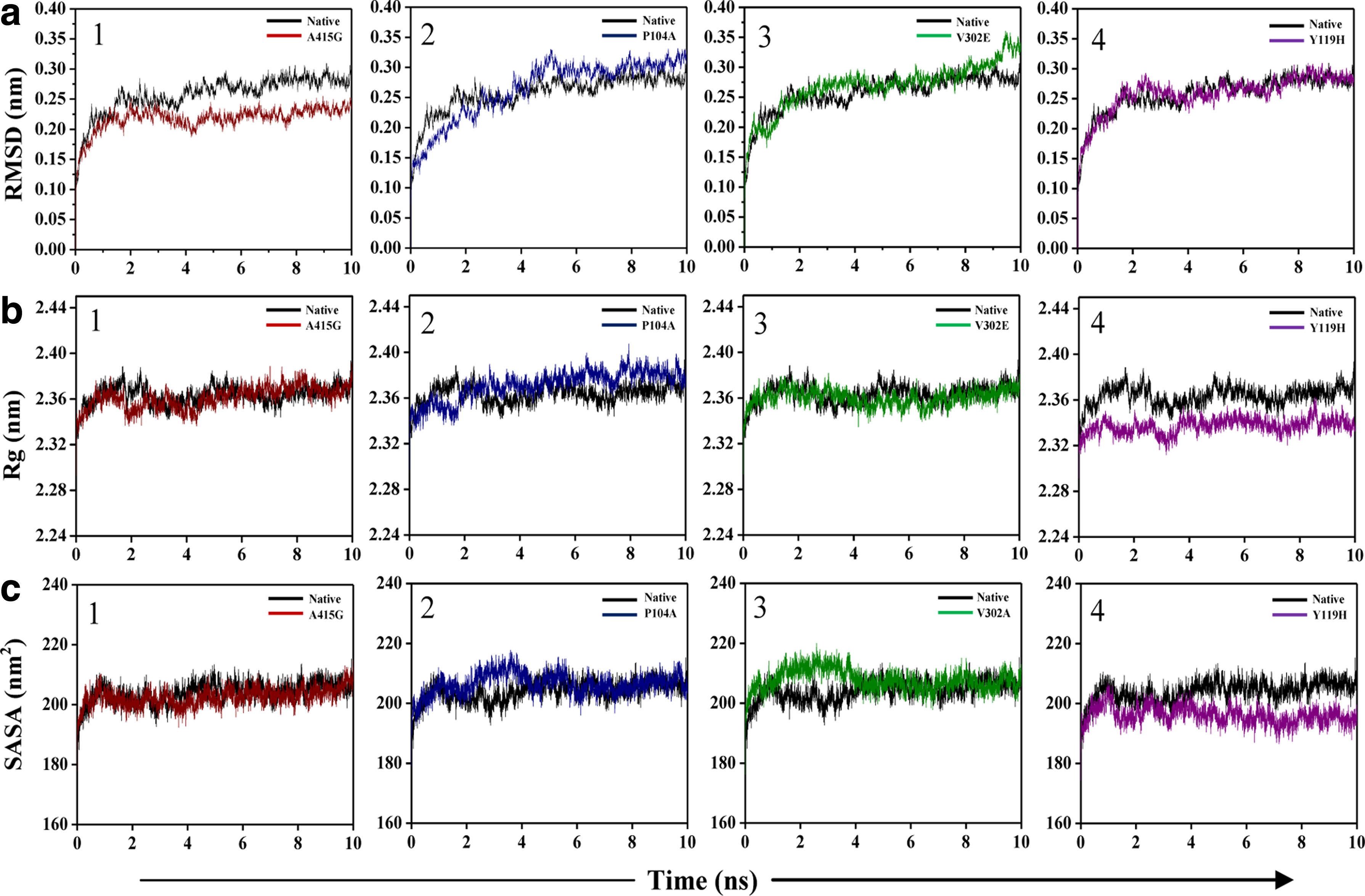
Structural properties of native and nsSNP forms of the hAChE protein. Protein backbone RMSD
Figure 3c shows the plot for solvent-accessible surface area (SASA) of nsSNP forms in comparison to the native. The SASA of A415G was almost similar to that of the native structure during the course of simulation. Though the plot of P104A and V302E initially had increasing patterns, after 4 ns both showed similar plot as that of the native. Y119H showed decreased SASA as in Rg, indicating the compactness of this nsSNP protein form during the course of simulation. Taken together, these results suggest that all these minor deviations in RMSD, Rg, or SASA may not impart any structural instability to the protein as the protein's integrity is maintained throughout the simulation.
3.6. Dynamical properties of nsSNP forms of hAChE
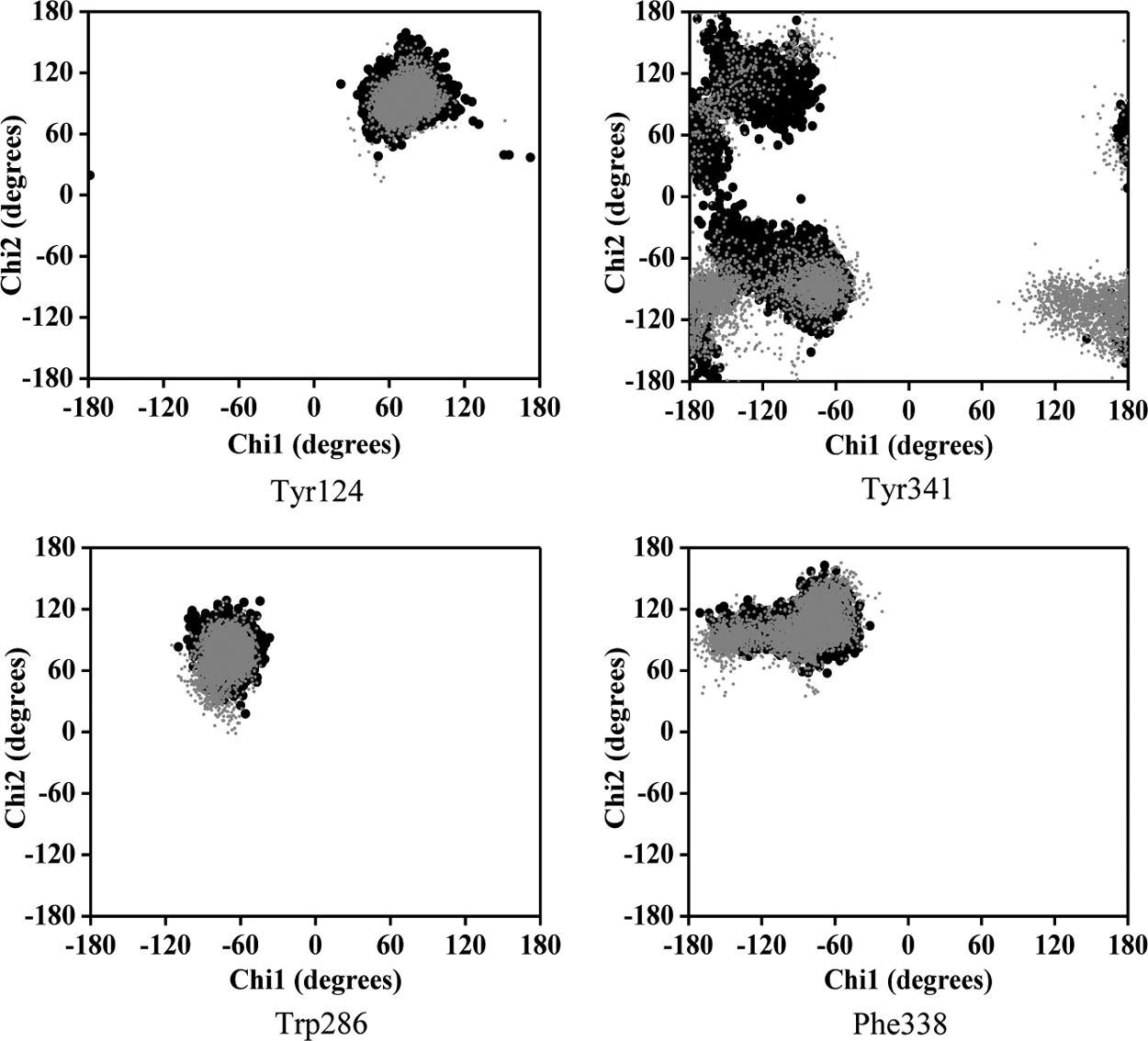
Xu et al. (2008) have elucidated the importance of 14 aromatic amino acids lining the active-site gorge (Fig. 4) in binding and trafficking of substrate and other ligands toward the active site. Among the 14 amino acid residues, we investigated the effect of mutations on the flexibility of PAS residues (Tyr124, Trp286, Tyr341, and Asp74) and bottleneck residue (Phe338) by measuring their side-chain dihedral angles (χ1 and χ2) throughout the 10 ns simulation.
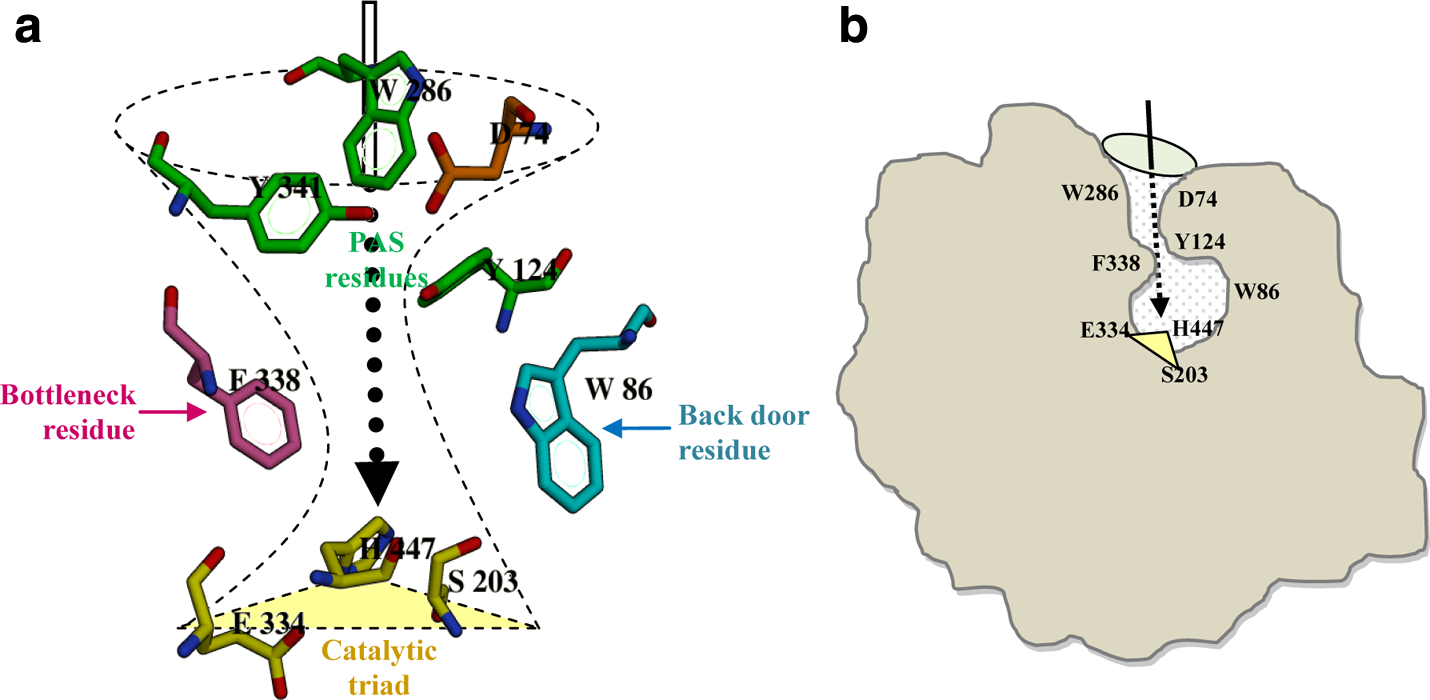
The orientation of aromatic amino acids along the active site of the hAChE protein.
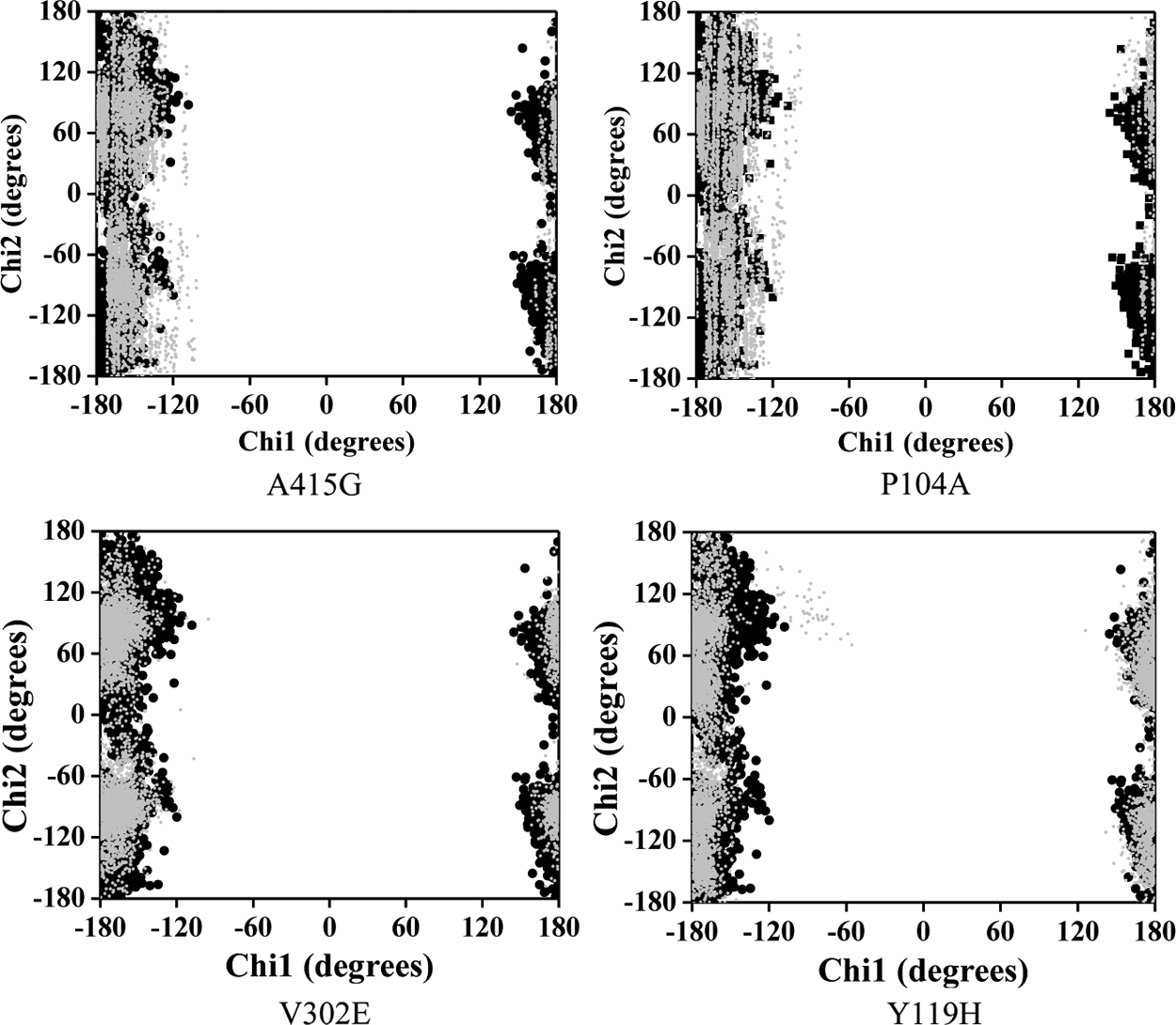
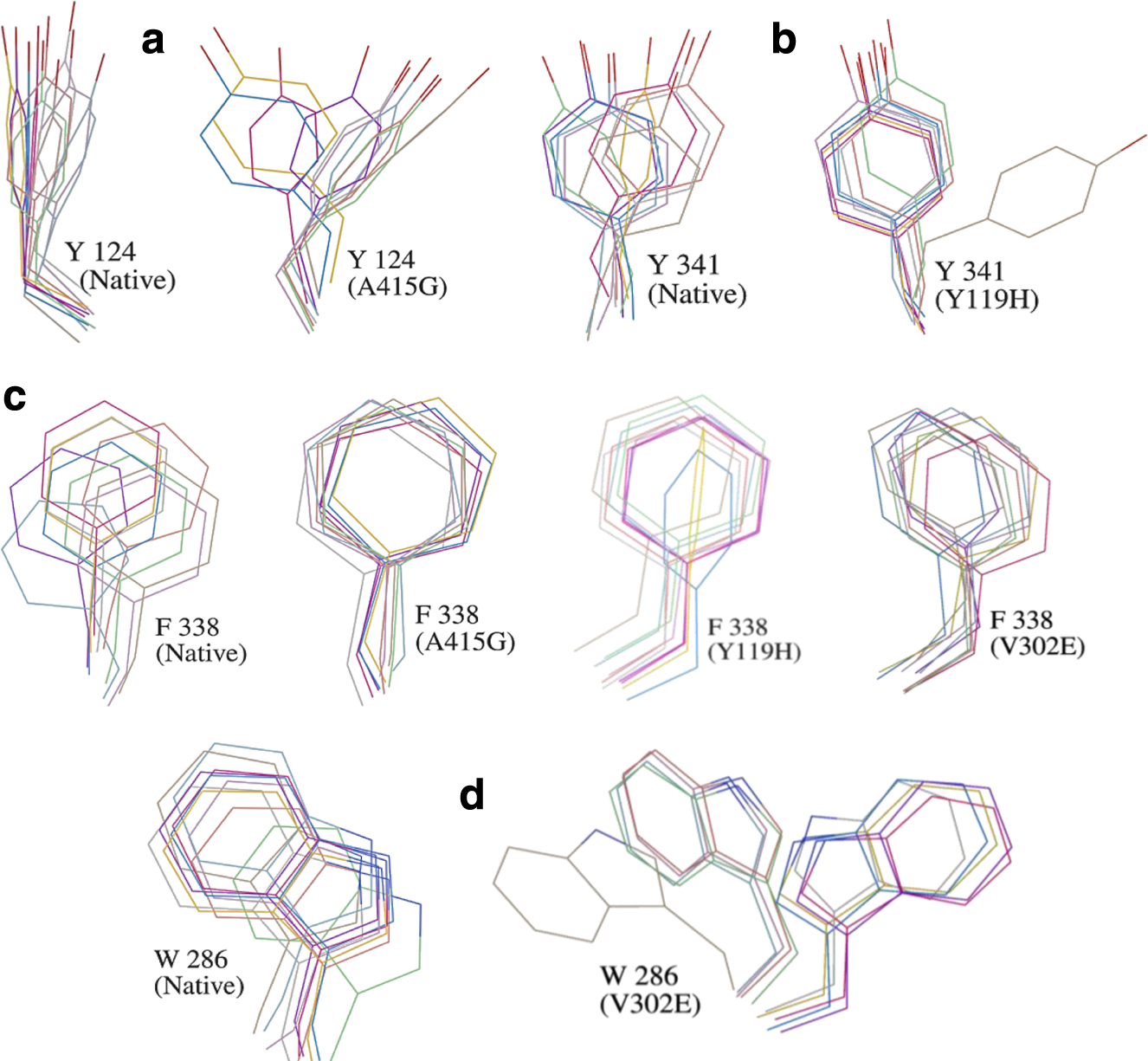
Analysis of the χ plot of PAS residues showed that the flexibilities of Tyr124 and Phe338 in A415G (Fig. 5a,b); Trp286 and Phe338 in V302E (Fig. 5c,d); and Tyr341and Phe338 in Y119H (Fig. 5e,f ) were affected. In A415G mutant, the χ1 angle of Tyr124 oscillates either at −60° or +60°, whereas in native, it was fixed around +60°. Similarly, Trp286 in the V302E form of the protein showed an oscillating pattern in its χ2 angle (−40° or +80°), which in native was fixed around +80°. In Y119H, the χ2 angle values of Tyr341 were widely distributed around −120°, while in native, it was clustered maximally around +100° and −80°. The χ1 angle of Tyr341 was also disturbed in Y119H. The χ plot of the bottleneck residue, Phe338, showed that the limited flexibility of the χ1 angle in native is further retained to a smaller area in A415G, V302E, and Y119H (Fig. 5b,d,f ). Moreover, Phe338 in Y119H showed a high degree of χ2 oscillation (either −60° or +130°), which in native was fixed at 100°. However, in P104A, not much conformational change was observed in any of the PAS residues and bottleneck residue (Fig. 6). Interestingly, the PAS residue Asp74 did not show any variation in any of the mutant forms studied (Fig. 7). As proposed from the dihedral angles, the change in side chain orientation of the key residues involved in ligand binding were distinctive on superimposing the backbone of mutant structures used as docking template onto the native (Fig. 8). Unlike other nsSNP forms, it is well noticeable that P104A did not show much variation, and therefore it is perceived that this mutation might not affect ligand binding.
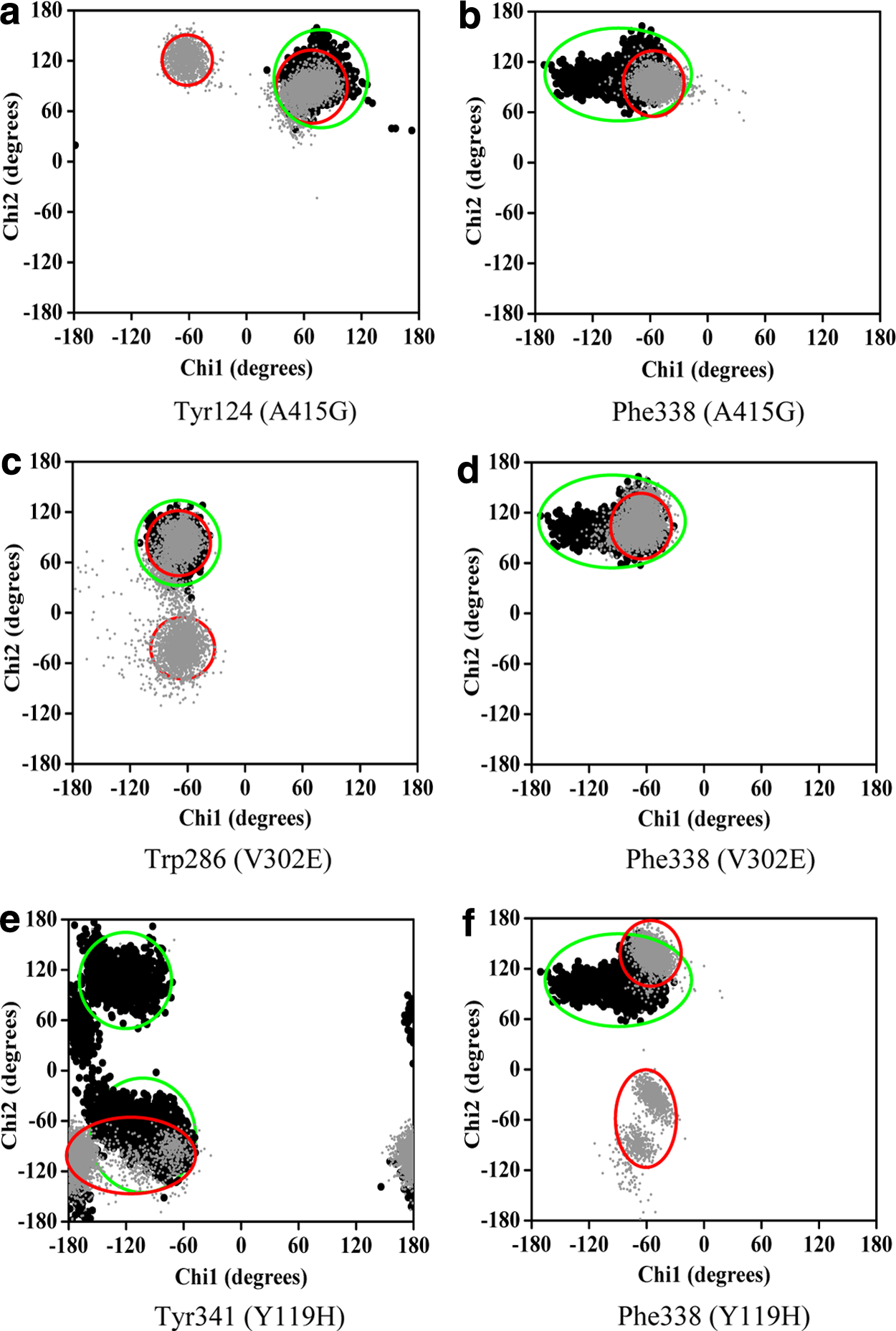
Dynamical property of the mutants in comparison with the native protein. Plots a–f show the χ1 and χ2 angles of amino acids lining the gorge, which reflect the flexibility of their side chains. The corresponding mutants are given in parentheses. Dark and gray data points represent native and mutant amino acid residues, respectively.
Dynamical property of the P104 mutant of hAChE. The χ plot of PAS and bottleneck residues in the P104A mutant of hAChE. Dark and gray data points represent native and mutant amino acid residues, respectively.
The side chain dihedral angles of Asp74 in all four mutants. The χ plot of the Asp74 residue in four different mutants of hAChE. Dark and gray data points represent native and mutant amino acid residues, respectively.
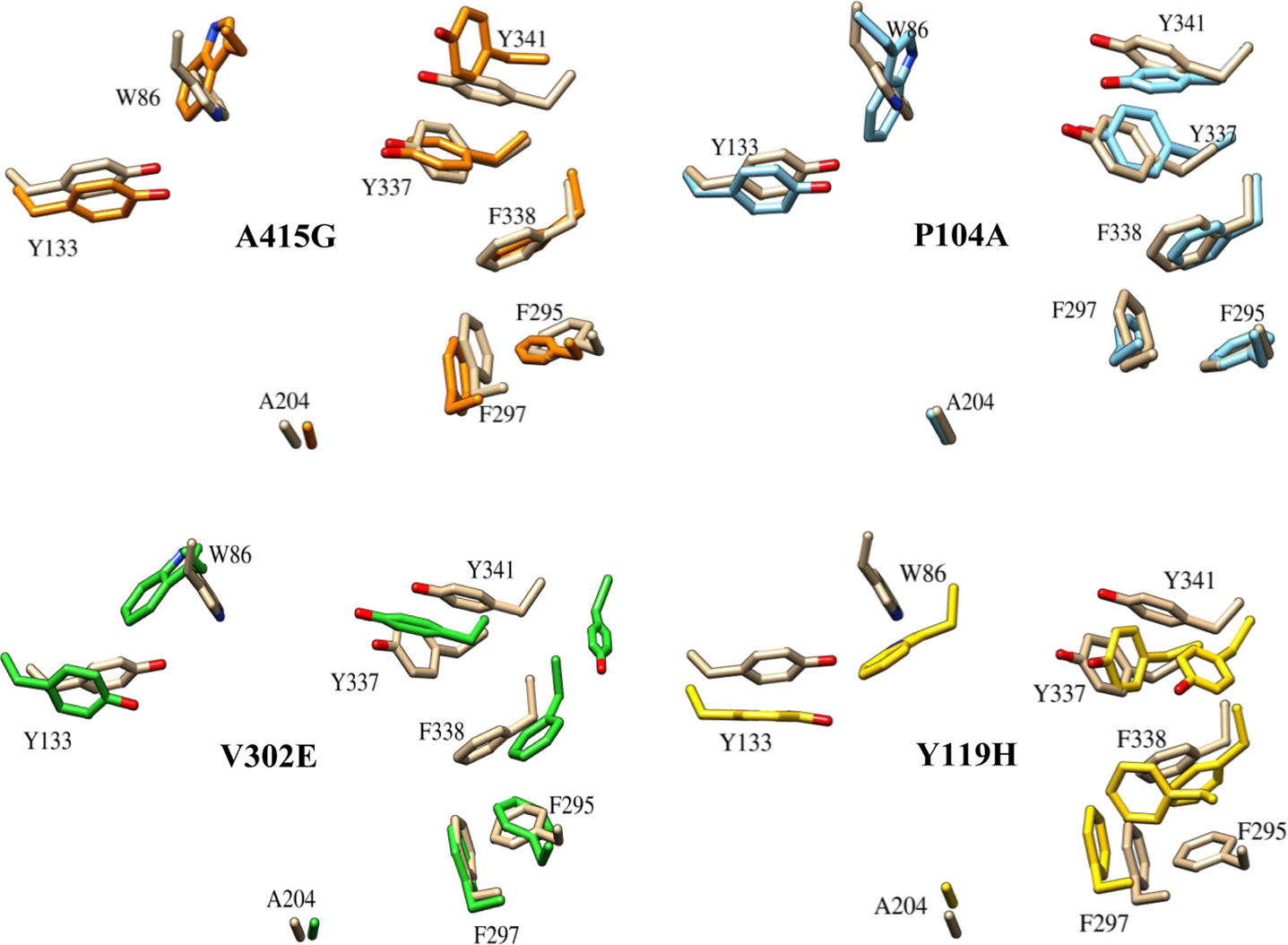
The orientation of active-site residues in four mutants. The figure shows the change in conformation of the key residues that play a vital role in binding of ligands. The positioning of the residues in four nsSNP forms (docking template) is shown with respect to the native, which is represented in brown.
Among the PAS residues, the indole ring of Trp286 has been implicated to form a stable protein–inhibitor complex through Π-cation or stacking interactions. Similarly, Asp74 is another PAS residue involved in electrostatic interactions with the ligand (Barak et al., 1994). Thus, the entry of substrate/inhibitors is gained through these two residues. In addition, substrate entry through gorge is also controlled by the gating action of the bottleneck residues. Among the bottleneck residues, the dynamicity of Phe338 is critical and its fluctuation could influence the binding of ligands. Tyr124, a PAS residue, is the standby second residue mediating the bottleneck obstructions (Shen et al., 2002). These residues are therefore indispensable for establishing a stable hAChE–ligand complex, and they show varied conformational flexibility in the nsSNP forms of hAChE. Any protein forms with such conformational variation are expected to have a high degree of variations in their binding pattern of hAChEI. Hence, docking studies were performed on these nsSNP forms for evaluating their binding efficiency with hAChEI that are widely used as therapeutic agents in AD.
3.7. Interaction analysis of hAChEI with hAChE
The docked structures of hAChE–ligand complexes generated in the present study were compared with already available hAChE–ligand crystal structure complex (4EY7, 4EY6, and 4EY5) to establish the binding mode of ligands under our study (Cheung et al., 2012). Figure 9 shows the reproducibility of ligand conformation from the PDB structures, and such standardized parameters were used to dock all the drugs onto mutant proteins. The top-ranking, low-energy conformants of hAChEI such as donepezil, galantamine, huperzine A, rivastigmine, and tacrine were deduced and analyzed for their binding efficiencies with all four nsSNP forms (Table 2). It was observed that although each of the ligands interacted differently with different mutants, none of them show any similarity to the interactions observed in the native–ligand complex. Particularly, the ligand donepezil in the complex of three mutants, namely, A415G, V302E, and Y119H, was bound at the PAS (Fig. 10). The explanation for this could be gained by observing the χ plot of amino acids at the PAS and bottleneck (Fig. 5) and their movement during the MD simulation (Fig. 11). In the A415G form of the protein, it is seen that the Tyr124 has a flip-flop kind of oscillation (Figs. 5a and 11a) and Phe338 has more restricted flexibility than the native (Figs. 5b and 11c). Both of these residues could offer steric hindrance to the drug, thus preventing it from reaching the active site (Fig. 10a). Similarly, in V302E mutant, Trp286 exhibited a wide fluctuation (Fig. 11d), which is in agreement with Figure 8b, where the drug is prevented from moving ahead of the residue. The Phe338 residue also showed lesser flexibility than the native (Fig. 11c). The flexibility of Tyr341 and Phe338 residues in native were drastically confined in the Y119H mutant (Fig. 11b,c), which has restricted the drug at the entrance of the gorge (Fig. 9c). However, the P104A mutant showed no difference in the binding pattern of donepezil (Fig. 9d) as the side-chain dihedral angles of the PAS and bottleneck residue were almost similar in orientation to the native form (Figs. 6 and 8). These results demonstrate that the positioning of hAChEI is governed by the orientation of the aromatic residues lining the gorge and that the high degree of their side chain fluctuations would affect the trafficking and orientation of the ligands for their interaction with catalytic site.
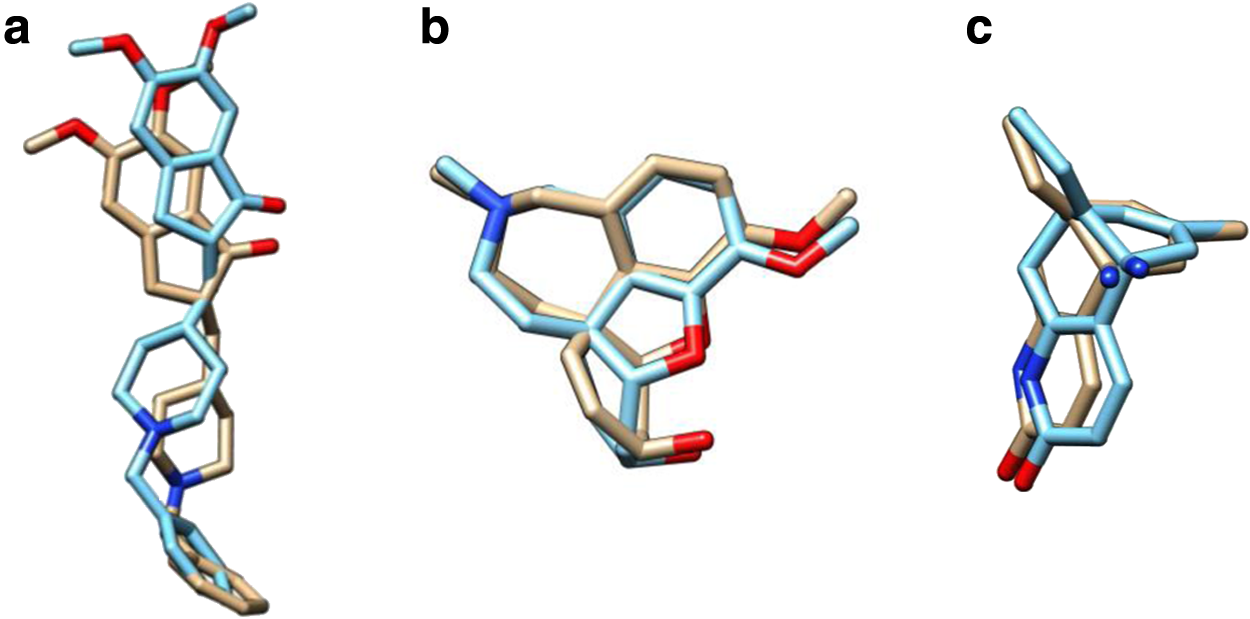
Superimposition of docked configuration of ligands on crystal structure. The ligands
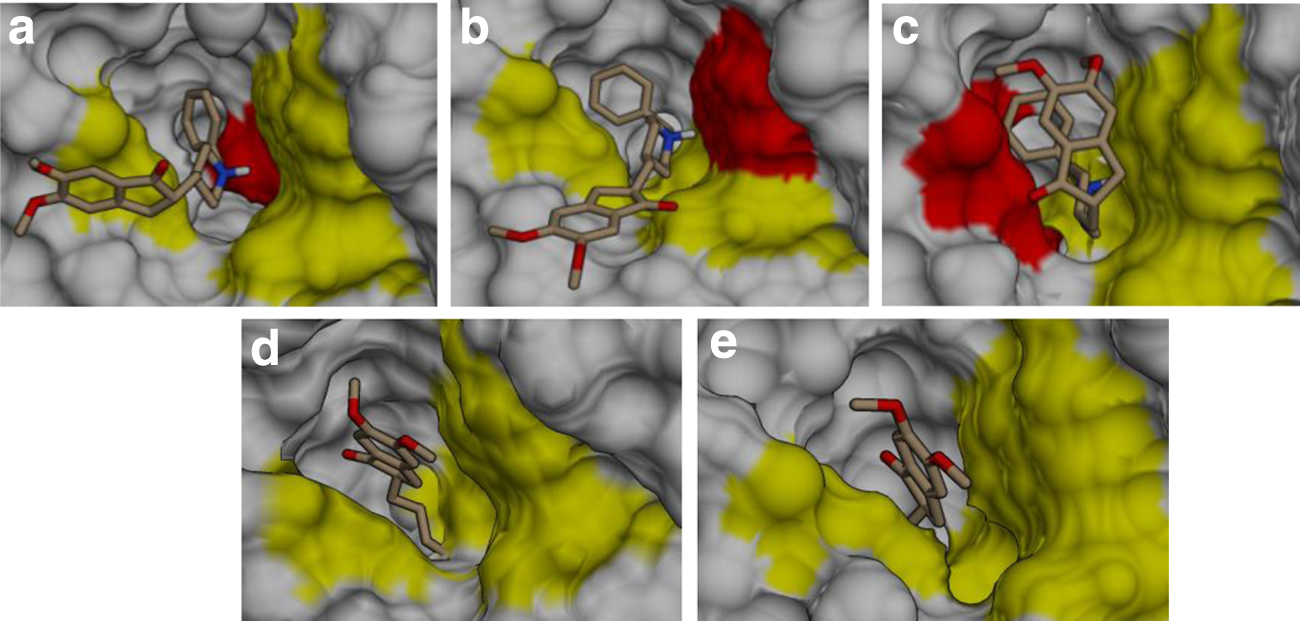
Binding orientation of donepezil with four nsSNP forms of hAChE. Docked conformation showing unfavorable binding patterns of donepezil with the four different mutants of the hAChE protein A415G
Superimposition of PAS residues at different time intervals during the course of molecular dynamics simulation. Movement of residues during the course of simulation in mutant forms of hAChE in comparison with the native
All other hAChEI, galantamine, huperzine A, rivastigmine, and tacrine with a smaller surface area than donepezil were able to tolerate the altered flexibility in the mutant protein molecule and thus could reach the active site with ease (Fig. 10b–e). However, galantamine, rivastigmine, and tacrine varied greatly in their orientation in comparison to the native, which is unfavorable for complete inhibition (Fig. 12a–c). From these figures, it is perceived that though the positioning of these ligands is identical to both native and mutant forms, the subsequent orientation of the ligand atoms widely differs from each other, which might be because of the altered dihedral angels of amino acids lining the active-site gorge. Interestingly, huperzine A had identical conformation in both the native and nsSNP forms (Fig. 12d). Additionally, from the binding energy it is seen that huperzine A has better binding affinity toward all the mutants than the native protein despite the fluctuations in the aromatic residues lining the gorge. The ability of huperzine A to have a higher affinity for hAChE irrespective of the nsSNPs could be because of Asp74. It is a key residue involved in its binding (Xu et al., 2003) and the χ angle of this residue was unaltered in all protein forms of hAChE (Fig. 7). Further, it is said that the PAS residues do not contribute to the stability of AChE–huperzine A complex (Ariel et al., 1998), from which we can further conclude that huperzine A might be a potent inhibitor irrespective of the altered fluctuations in any PAS residues of several polymorphic forms of hAChE. Donepezil and Huperzine A inhibit hAChE at lower concentrations (IC50: 22 and 47 nM) than other AD drugs (Luo et al., 2005), but donepezil differs in the binding conformation within the active site of the mutant forms. Fortunately, huperzine A displays proper orientation at the active site of mutant proteins, and hence huperzine A can be considered as the best drug against hAChE. Moreover, huperzine A's potency and acceptability in patients (Rafii et al., 2011) reflect its safety and its possible wider use as a drug of choice in the treatment of AD.

Overlay of the docked conformations of drugs. The conformation of drugs galantamine
Taken together, these results emphasize that for an effective therapeutics against any disease, the genetic constituent of an individual, especially the common, nonpathogenic nsSNPs, should be investigated. This study was carried out to understand the basis of intolerance exhibited by the population with respect to nsSNPs. From our results, we conclude that the present-day drugs are highly specific to protein conformation and become ineffective because of the change in the orientation of active amino acid residues conferred by the nsSNPs of the protein. Second, our study suggests that huperzine A would be the best option as a first-line therapy for AD, as it is the only drug that has higher efficiency in its binding to the hAChE protein irrespective of its polymorphisms. Therefore, considering the diverse polymorphic nature of the hAChE protein, while contemplating any structure-based drug designing, the common, nonpathogenic nsSNPs should be considered in evolving the best drug.
4. Conclusion
Considering the AD treatment, the need for understanding an individual's response to a drug and alternative drugs with higher hAChE inhibitory efficiencies to replace the current FDA-approved hAChEI exists. The present MD and docking study shows that various nsSNP forms exhibit different dynamic properties that affect the inhibitors' interaction, either in its orientation or in its binding patterns. Although the four nsSNPs (A415G, P104A, V302E, and Y119H) that were predicted to be functionally unfavorable showed least structural instability, their aromatic amino acids lining the gorge occupied alternative conformations, which in turn affected the drug trafficking, resulting in ineffective drug orientation at the active site of hAChE. This could be the reason for differential effect of FDA-approved hAChEI on AD patients. However, huperzine A demonstrated a better affinity toward all the four nsSNPs of hAChE than any other known AD drug. Thus, this study highlights that the individualized varied response observed with AD drug is precisely because of nsSNPs of the hAChE protein. Second, we propose that the therapy targeting the hAChE protein, which is a major component of the cholinergic system, could now be channeled toward usage of huperzine A as the first-line treatment in AD.
Footnotes
Acknowledgments
P.S. and R.K.C., respectively, acknowledge the research fellowships DST-INSPIRE and DST-PURSE, both provided by the Department of Science and Technology, New Delhi.
Author Disclosure Statement
The authors declare that no competing financial interests exist.