
Abstract
Abstract
1. Introduction
T
Constraint-based methods have proven to be very successful in the analysis of metabolic networks (Papin et al., 2004; Price et al., 2004). In constraint-based methods no detailed information on reaction kinetics is needed. Often, the knowledge of reaction stoichiometries is sufficient. Rows of the stoichiometric matrix correspond to metabolites and columns to reactions: sij the i,j-th entry of S is the number of molecules of compound i consumed (sij < 0), produced (sij > 0), or not involved (sij = 0) in reaction j. In steady state this results in linear constraints that express flow conservation on all internal metabolites, with possibly lower and upper bound on fluxes, yielding a polyhedron of feasible flux-vectors P = {v : Sv = 0,ℓ ≤ v ≤ u}.
Among the most prominent analysis methods is flux balance analysis (FBA) (Varma and Palsson, 1994; Orth et al., 2010; Santos et al., 2011). It is, for example, used to compute the optimal biomass yield that can be achieved by a cell under some growth medium (Feist and Palsson, 2010). This amounts to solving a linear programming problem of the form max{cv : v ∈ P}, where cv is the linear function expressing the (weighted) amount of biomass. In general such optimal flows are not unique (Mahadevan and Schilling, 2003). If this is ignored, it can lead to wrong predictions of byproduct flux rates (Khannapho et al., 2008).
Kelk et al. (2012) showed that many reactions have fixed flux rate in all optimal solutions. These are determined by flux variability analysis (FVA) (Burgard et al., 2001; Mahadevan and Schilling, 2003). The remaining variability is due to variability of the fluxes on a number of relatively small subnetworks, which we call flux modules. As an example, we use here an artificial network similar to the one presented by Müller and Bockmayr (2013b) in Figure 1.
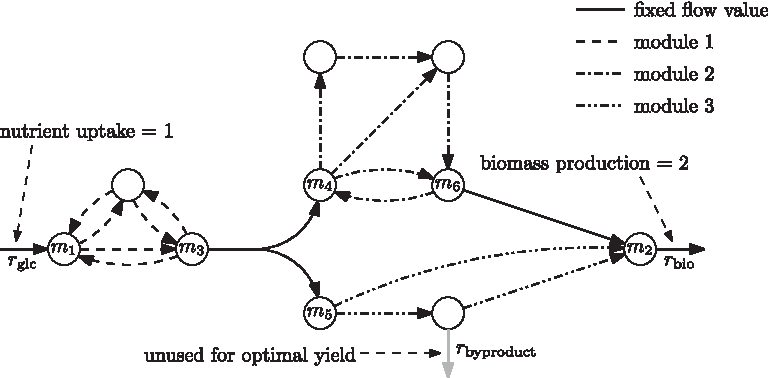
Toy example network. All stoichiometric coefficients are +1, −1, or 0. We assume a fixed nutrient uptake rate of one through rglc. For optimal biomass production (flux through rbio) this implies that no side product is produced (flux through rbyproduct is fixed to zero) and an optimal biomass production of two is achieved. The continuous hyperarcs represent reactions carrying fixed flux in all optimal solutions. The modules of the network are marked with different dash styles. Since the reaction rbyproduct carries no flux in any optimal solution, it is grayed out.
In the example, all stoichiometric coefficients are supposed to be +1, −1, or 0. Assuming an input flux rate 1 of the nutrient, any optimum outputs rate 2 of biomass. The continuous hyperarcs represent the reactions that carry fixed flux in any optimal solution. The various dashed sub-hypernetworks indicate the variability present in various optimal solutions. However, for any of the dashed, dash-dotted, or dash-dot-dotted, hypernetworks, we notice that the net influx and the net outflux is the same in every optimal solution: for example in every optimal solution, the dash-dotted subnetwork consumes one unit of metabolite m4 and produces one unit of metabolite m6, but there is flexibility in which route this unit flow goes through the dash-dotted network. This allows for seeing a module as one sort of aggregated reaction (see Fig. 2).
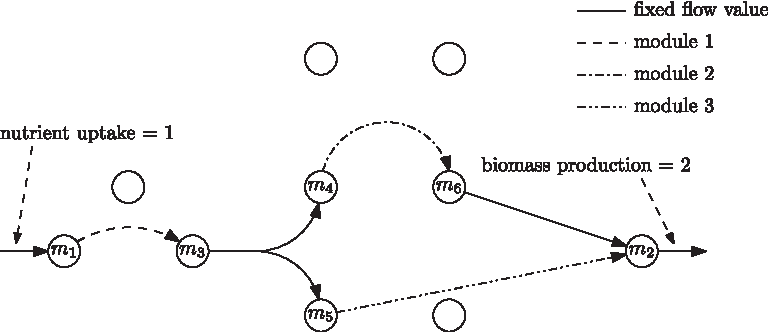
The modules replaced by aggregated reactions.
The fixed input and output compounds of a subnetwork characterizes the notion of flux-module (Müller and Bockmayr, 2013b) in a mathematically rigorous way. Müller and Bockmayr (2013b) showed that every optimal yield elementary flux mode (EFM) (Schuster and Hilgetag, 1994) is a concatenation of reactions with fixed flux and an elementary mode of each of the flux modules. This is illustrated in Figure 3. There are two ways to go through the dashed module, three ways to go through the dash-dotted module, and two ways to go through the dash-dot-dotted module. Hence, 7 subpaths suffice to define the 12 elementary optimal flux modes. Clearly in large networks this combinatorial explosion can be much more dramatic.
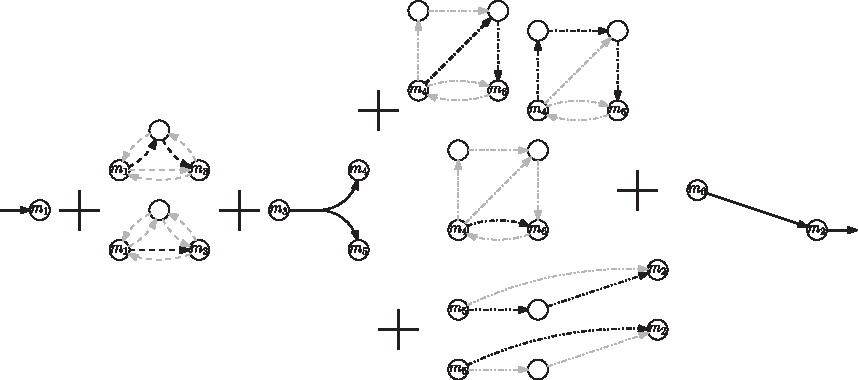
Visualization of all 12 optimal-yield EFMs of the toy network (Fig. 2). By taking one EFM through each module, together with the reactions of fixed nonzero flux, we obtain an optimal yield EFM of the original network. Furthermore, all optimal yield EFMs of the original network can be obtained this way. For each EFM of a module, the used reactions are marked in black, while the unused are marked in gray.
While the method by Kelk et al. (2012) required the enumeration of exponentially many vertices of a flux polyhedron (which are related to the optimal yield EFMs), Müller and Bockmayr (2013b) showed a way to find the modules without needing to compute all extreme solutions. Their method however relied on many runs of FVA. Although faster than EFM enumeration, the method is very sensitive to numerical instabilities, and analyses of genome-scale networks could still take several hours.
The most important result in this article is an extremely simple method allowing to compute the flux-modules in a few seconds for genome-scale metabolic networks. The method, described in section 2, is based on the observation that the modules correspond to the separators of the linear matroid defined by the columns of the stoichiometric matrix that belong to reactions with variable optimal flux. We will explain all these technical concepts in section 2.1. The efficiency of our method is demonstrated in section 3 by application to several genome-scale metabolic networks.
Flux modularity highly depends on the growth conditions. In particular, interesting flux modules can usually only be found in the optimal flux space. Hence, it is of high importance to understand how the decomposition of modules changes under different growth conditions and objective functions. Since with our new method, module computation has become so fast, we can simply compute and compare modules under many different growth conditions and compare the results. Essential for this is a visualization method that shows the interplay of modules in the context of the whole network. In section 2.3 we present a method that automatically generates such a visualization using a clever compression based on flux modules. Results of that method applied to a set of genome-scale metabolic networks can be found in section 3.2.
2. Methods
2.1. Definitions and preliminaries
We use
We will also be interested in the flux through a subset of reactions
In contrast to the definition in Müller and Bockmayr (2013b), we also allow A = ∅ to be a module, which together with
(i) If disjoint sets A and B are P-modules then A ∪ B is a P-module;
(ii) If A and B are P-modules and B ⊂ A then A\B is a P-module.
The rest of this section is devoted to an introduction to the relevant concepts from Matroid Theory (Oxley, 2011), which is a generalization of graph theory and linear algebra. A matroid is defined by a universe of elements and subsets of them that have some independence structure.
•
• If
• If
As a very relevant example, a set of vectors in
Matroid theory inherits many powerful concepts from linear algebra like duality and rank (which is important for the proofs displayed in the Supplementary Material, available online at www.liebertpub.com/cmb). Also graph theory introduces some further useful concepts into matroid theory. Important for us is the notion of a connected component of a matroid: two elements of a matroid are in the same component if there exists a circuit that contains both. We notice that in graph theory this property characterizes a two-connected component. A separator of the matroid is now any union of connected components, that is, any of the two sides of a partition of the matroid into two parts A,B such that there exists no circuit intersecting A and B. In section 2.2 we show how the flux modules of a metabolic network correspond one-to-one to the separators of the corresponding matroid. We then use matroid theory to derive a very fast and simple algorithm for finding modules. It is based on a result by Krogdahl (1977). The runtime results on a set of genome-scale metabolic networks are presented in section 3.1.
2.2. Finding modules efficiently
We first show that it is sufficient to analyze modularity as a local property of one point in the inside of the flux space, implying that we can ignore reaction reversibilities and simply analyze a subvector space (Theorem 1). This allows us to describe modularity in terms of matroid separators (Theorem 2), which we then exploit in designing an efficient algorithm to compute modules.
To make the first step, consider a point x inside the flux space and a neighborhood of it (Fig. 4).
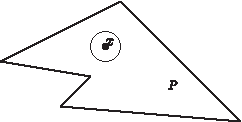
Viewed from a point x inside the flux space, the flux space looks like a linear vector space, and the bounds are not important.
This neighborhood captures all the characteristics needed to analyze modularity of the whole flux space. We only have to deal with the term “inside.” Since
This restriction does not destroy the module property:
To guarantee that we can find an x inside the flux space after we restricted to reactions with variable flux rate, we require that P is convex. In a future work we will consider the case of nonconvex flux spaces.
A is P-module ⇔ A ∩ V is ker(SV )-module.
The proof can be found in the Supplementary Material.
By Theorem 1 we can restrict our attention to the analysis of linear vector spaces. Hence, in the following we will only analyze polyhedra of the form P = ker(S). We will relate modules of ker(S) to separators of the matroid defined by the columns of S. Remember the explanation of a separator in a graph in terms of the nonexistence of a flow circulation in section 2.1 and observe that every module in ker(S) also has interface flux 0 since
Formally, we obtain the following theorem, the proof of which is deferred to the Supplementary Material.
The characterization of modules as separators of matroids allows to compute the flux-modules of a metabolic network efficiently. Since separators and modules are closed under disjoint union, it suffices to describe the set of minimal nontrivial separators (modules).
To understand the algorithm for finding the modules, we observe that the minimal nontrivial separators are the connected components of the matroid. Formulated in matroid-terminology we recall from section 2.1 the following characterization of connected components For any two elements (columns of S in the linear matroid, edges in the graph) in the same connected component there exists a circuit that contains them both. For pairs of elements of different connected components this is not true.
We can now build a graph G = (V,E), where V is the set of reactions defined in (1) and there is an edge between two reactions (columns of SV) if and only if there exists a circuit that contains both. The connected components (in the graph-theoretic sense) of G will be the minimal separators. However, as the number of circuits explodes exponentially, it is not efficient to enumerate all circuits in order to compute the connected components of the graph G. Indeed, this is also not necessary and it suffices to look at a special set of circuits, so-called fundamental circuits (Truemper, 1984).
A set of fundamental circuits is obtained as follows: We start by finding a maximal independent set (also called basis) X of the matroid, which we compute by Algorithm 1. Notice that, starting from the empty set, the algorithm grows X by adding elements only if this keeps X independent. Since we try to add all elements to X, it follows that at the end of the algorithm, X will be a basis of the linear matroid represented by SV.
Let Y := V \X. Clearly, for every
We now build, by Algorithm 2, the graph G′ = (V,E′), where two reactions are connected by an edge if there exists a fundamental circuit that contains both. Krogdahl (1977) and Cunningham (1973) showed that the connected components of G′, found by Algorithm 2, are precisely the minimal separators of the matroid.
To each circuit C there exists a flux vector v that is unique up to scaling with C = supp(v), Sv = 0. If we enter for every fundamental circuit the corresponding flux vector as a column into a matrix, we obtain a null-space matrix of S. Hence, this approach can be understood as computing a block-diagonalization of the null-space matrix. Approaches like this in the context of stoichiometric matrices have already been studied in Schuster and Schuster (1991). However, Schuster and Schuster (1991) do not use matroid theory, and it is unclear whether their method will always compute the finest block-diagonalization.
Computes a basis X and its set of fundamental circuits of a matroid represented by S
Computes the modules of {v : SV v = 0}
Here we recapitulate all the steps for finding the modules of the optimal flux space of a metabolic network.
1. Determine the optimal value by LP;
2. Set the objective function equal to the optimum value and add it as a constraint;
3. For each reaction r maximize and minimize the flux through r in the optimal flux space;
4. Determine the set V of reactions for which the maximum and the minimum are not equal;
5. Select the set of columns SV corresponding to V of the stoichiometric matrix S and neglect the non-negativity constraints, i.e., irreversibilities, directions of the reactions;
6. Apply Algorithm 2 to compute the minimal modules
7.
We notice that step 3 (and therefore 4) of the algorithm can be parallelized in a trivial way, reducing the computation times even further.
2.3. Visualization
We develop a visualization tool to help us understand how the decomposition of modules changes under different growth conditions and objective functions. By the definition of module, the reactions inside a module have together a fixed function (the interface flux). Hence, we can represent the module by a single reaction with a fixed flux in the genome-scale network. The stoichiometry of the representing reaction is precisely the interface flux of the module.
This way we can create a compressed network that contains all the reactions with fixed flux rates and artificial reactions that represent the modules. This compressed network has the following advantages:
• The number of reactions carrying flux is compressed (a module with many reactions is represented by a single reaction). • All the reactions in the compressed network have a fixed flux rate.
Unfortunately, the number of fixed reactions is still very large. This prevents automatic visualization of the network and the role of the modules containing variable reactions is obfuscated. However, reactions that have a fixed flux rate can also be grouped together into modules by Proposition 1.
Theoretically, we could group all reactions with a fixed flux rate into one module. This would result in a compressed metabolic network consisting of k + 1 reactions, where k is the number of minimal modules containing reactions with variable flux rates. In particular, the module containing all fixed reactions will likely also contain the biomass- and nutrient-uptake reactions. If we want to understand the role of the modules for biomass production or nutrient uptake, this is not very useful. Moreover, modules of variable reactions may disconnect reactions with fixed flux rates from each other. Such disconnected reactions are important for the mediation between modules and should also be displayed separately. Hence, we decided to build a compressed network as follows:
1. Given: A collection Mod of interesting modules (selected by the user). Mod has to cover all reactions with variable flux rates. Typically Mod contains all minimal modules of variable reactions, a module containing the biomass reaction and modules containing the nutrient uptake reactions. 2. We compute the set 3. We compute the set 4. We compute the set 5. We consider the metabolic network, where 6. We compute the connected components ModF of S′. We do so by defining the incidence matrix of a bipartite graph, the nodes of which on one side of the bipartition correspond to the rows of S′, and the ones on the other side to the columns of S′, and there is an edge between row-node i and column-node j if and only if 7. We represent each module in Mod, ModF by a single reaction with the corresponding interface flux. Let 8. We remove reactions disconnected from the network that contain the target reaction, for examples because of modules that form thermodynamically infeasible cycles or otherwise have no role in the metabolism.
In practice, this results in medium-scale networks that can automatically be visualized with graph-drawing software like GraphViz (Gansner and North, 2000).
3. Results
3.1. Runtime of module finding
With the new method we can compute all flux modules for the optimal flux space of genome scale networks in about the same time as is needed for conventional flux variability analysis. In Table 1 we see that the new method using matroid theory outperforms the previous methods in orders of magnitude. We used the metaopt toolbox (Müller and Bockmayr, 2013a) to solve the flux variability subproblems. Unfortunately, we did not have access to all the runtime data of Kelk et al. (2012), which is why some of the data is missing and the reported runtimes may be only from some steps in the pipeline. The computations for the matroid approach were obtained by computations on a four-core desktop computer.
In particular, notice that large networks like Human recon 2 can now also be analyzed. In addition, the new method is numerically much more stable. In the method introduced by Müller and Bockmayr (2013b) it often happens that error tolerances are chosen too small or too large, which causes linear programs that should be feasible to be detected as infeasible, etc. This then usually caused the algorithm to abort and the tolerance sometimes needed to be adjusted according to the problem instance.
We experienced that the new matroid-based method is much more robust in this respect. Our initial tolerances of 10−20 for the optimization step, 10−8 for the flux variability, and 10−9 for the final module computation worked in all cases.
Note that the other two methods are solving slightly different problems. In Müller and Bockmayr (2013b) we were actually looking for modules in the thermodynamically constrained flux space, and in Kelk et al. (2012), rays and linealities are eliminated prior to module computation.
A comparison between the results of Müller and Bockmayr (2013b) and the new method on E. coli iAF1260 revealed that seven of the modules coincide and two modules from the new method contain additional reactions (which have fixed flux under thermodynamic constraints). The remaining modules are computed by the new method but not by Müller and Bockmayr (2013b) since they again only contain reactions that have fixed flux by thermodynamic constraints (usually those modules are formed by a split pair of forward and backward reactions). The differences seem to be small, but a detailed analysis will be subject to future work.
3.2. Visualization
We used the visualization method presented in section 2.3 to create visualizations of the above-mentioned genome scale networks. The results can be found online. In Table 2, we compare the original size of the networks with the size of the compressed networks that are used to visualize the interplay of the flux modules with variable flux rates. Each reaction of the compressed network is a flux module. Every minimal flux module containing reactions with variable flux rates is represented by exactly one reaction. Reactions with fixed flux rate are grouped together. It is interesting to see that although the networks have quite different sizes originally, the compressed sizes do not vary very much.
For each of the genome-scale networks a compressed network representing the optimal-yield flux space was computed by compressing flux-modules and sets of reactions with fixed flux to single reactions. All reactions in the compressed network have a unique flux, and the metabolites display the interactions between the flux modules.
Visualizations of some of the example networks and their modules, using the tool dot (Gansner et al., 1993) from the GraphViz toolbox, can be found online. The MATLAB scripts for module detection can be found online as well.
4. Discussion
4.1. Enumeration of optimal-yield pathways
We showed that flux modules (Kelk et al., 2012; Müller and Bockmayr, 2013b) of genome-scale metabolic networks can be computed efficiently using matroids. We confirmed the previous results that the optimal flux space of most genome-scale metabolic networks decomposes into modules. If we want to compute the set of all optimal yield elementary modes, we theoretically can do this by simply computing the optimal yield elementary modes for each module. Then, we can use the decomposition theorem of Müller and Bockmayr (2013b) and obtain all optimal yield elementary modes of the whole network. A small numerical barrier in practice is that EFM enumeration for each module appears to be numerically very unstable. Hence, it is likely that EFMs are missed if not everything is computed using precise rational arithmetic.
We noted that the previous methods (Kelk et al., 2012; Müller and Bockmayr, 2013b) were computing flux modules on slightly different flux spaces [in Kelk et al. (2012) rays and linealities were removed; in Müller and Bockmayr (2013b) we worked on the thermodynamically feasible flux space)]. These differences seem to be small but could be of significant biological importance. For example, it could be that due to thermodynamic constraints a reaction is blocked and hence, we can refine the modules. In a follow-up work we will (mathematically and empirically) analyze the impact of these differences. Also, we want to point out here that, for computing modules, the method by Kelk et al. (2012) has to enumerate all the extreme points of the flux polyhedron of optimal fluxes (after some preprocessing), a much harder task. As a result more information than modules is obtained.
The full flux space is usually not decomposable into modules. In a follow-up article we will generalize the notion of module. This will allow us to find interesting modules also for the full flux space. Furthermore, this will have the potential to derive similar decomposition theorems as in Müller and Bockmayr (2013b) that then will work on the full flux space as well. We think this will be a major step toward EFM enumeration of genome-scale networks.
4.2. Modularity under different growth conditions
It has been observed that the decomposition into modules depends on the growth condition (Kelk et al., 2012; Müller and Bockmayr, 2013b). If we want to understand how the optimal flux space changes if the growth condition is modified, we have to recompute the decomposition into modules. Previously, this was a tedious task. Now it is very simple and fast, and it can be done even for very small changes.
We presented a visualization method that shows the interplay of the modules and how they contribute to optimal biomass production. We think that this visualization will be very helpful to detect when a change in a growth condition significantly changes the structure of the optimal flux space.
For the visualization we use the definition of module to lump reactions together. This way we compute a compressed metabolic network that shows the optimal flux distribution with only a small number of reactions. These networks were small enough to be visualized using automated graph-drawing tools. Currently, we have only little control on how these networks are drawn, causing the visualization to seem to be very sensitive to changes. In particular, it would be interesting if we could get more robust drawing results for small changes in the network.
4.3. Conclusions
In this article we demonstrated the power of matroid theory for metabolic network analysis and used it to present a new method that allows us to compute flux modules very efficiently. This allows us to compute flux modules of many metabolic networks under a large set of different conditions to compare flux modules with existing classical metabolic subsystems like glycolysis.
Compared to classical metabolic subsystems that, at worst, are arbitrary functional groupings of metabolic reactions/species, flux modules are mathematically well defined. They are structural features only depending on a defined set of conditions (inputs, optimality). This qualifies them as a performance and quality metric for genome-scale metabolic networks. Furthermore, it allows us to investigate the modularity, and simplify genome metabolic networks without the risk of a bias from conventional biological interpretation.
Footnotes
Acknowledgments
Author Disclosure Statement
No competing financial interests exist.
Authors' Contributions
A.R. and L.S. found the connection to matroid theory and took care of mathematical correctness. A.R. provided all detailed proofs. L.S. supervised and organized the collaboration process. A.R., F.B., and B.O. developed the visualization method. A.R., L.S., and F.B. worked on the manuscript. All authors read and approved the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.