
Abstract
Abstract
We introduce PASTA, a new multiple sequence alignment algorithm. PASTA uses a new technique to produce an alignment given a guide tree that enables it to be both highly scalable and very accurate. We present a study on biological and simulated data with up to 200,000 sequences, showing that PASTA produces highly accurate alignments, improving on the accuracy and scalability of the leading alignment methods (including SATé). We also show that trees estimated on PASTA alignments are highly accurate—slightly better than SATé trees, but with substantial improvements relative to other methods. Finally, PASTA is faster than SATé, highly parallelizable, and requires relatively little memory.
1. Introduction and Motivation
M
We present PASTA, practical alignments using SATé and TrAnsitivity. PASTA begins with an alignment and tree estimated using a very simple profile HMM-based technique and then realigns the sequences using the tree. If desired, a new tree can be estimated on the new alignment, and the algorithm can iterate. We demonstrate PASTA's speed and accuracy on a collection of biological and simulated datasets, including a 200K-sequence RNASim dataset (Guo et al., 2009), which we align in less than 24 hours using PASTA on a 12-core machine.
2. PASTA
PASTA uses an iterative strategy, and each iteration involves six steps (Fig. 1). The first iteration begins with the starting tree, and subsequent iterations begin with the tree estimated in the previous iteration. In each step, the guide tree is used to divide the set of sequences into smaller subsets, and to build a spanning tree with these subsets as nodes. We independently estimate MSAs for all the sequence subsets. Then, pairs of MSAs corresponding to subset that are adjacent in the spanning tree are aligned together using existing tools that can align MSAs. The resulting collection of MSAs overlap each other and are compatible where they overlap. These properties enable us to merge these overlapping MSAs using transitivity and generate an MSA on the entire set of sequences. Finally, a maximum likelihood tree is estimated on the final alignment. We provide a description of these steps in their default settings, but see Appendix C.1 in the Supplementary Material (available online at www.liebertpub.com/cmb) for full details.
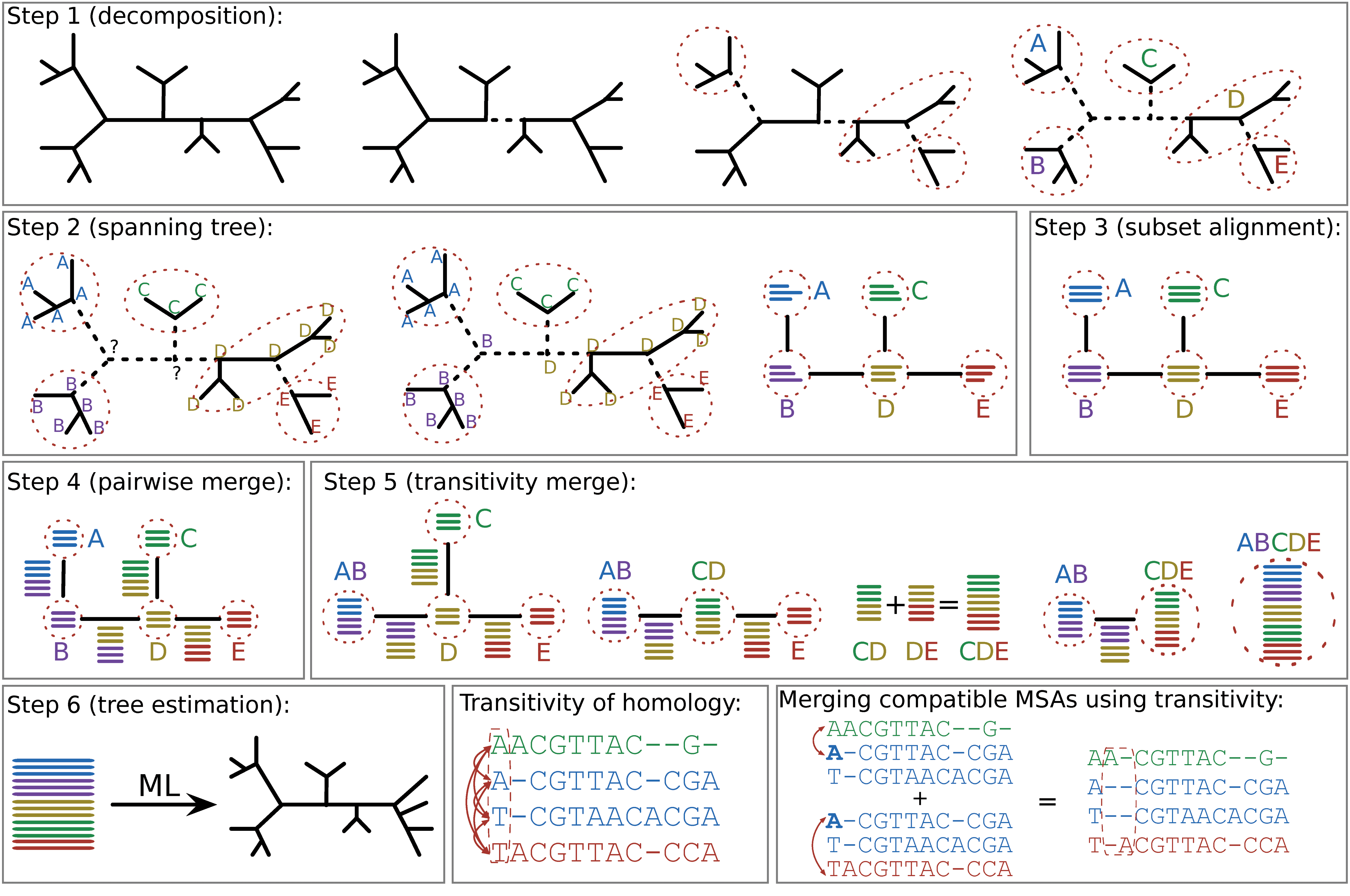
Algorithmic design of PASTA. The first six boxes show the steps involved in one iteration of PASTA. The last two boxes show the meaning of transitivity for homologies defined by a column of an MSA, and how the concept of transitivity can be used to merge two compatible and overlapping alignments. MSA, multiple sequence alignment.
Proofs have been omitted due to space constraints (see Appendix C.2 in the Supplementary Material).
3. Experimental Setup
3.1. Datasets
All the datasets used in this study are available online (See Appendix F in the Supplementary Material for download links).
To explore performance on larger datasets, we simulated 10,000-sequence datasets using Indelible v. 1.03 (Fletcher and Yang, 2009) under three different rates of evolution (10 replicates each), with average sequence length 1000.
To explore performance on ultra-large datasets (up to 200,000 sequences), we subsampled from the million-sequence RNASim (Guo et al., 2009) dataset (available online), with average sequence length 1556. RNASim is a simulator for RNA sequence evolution that we present here, and that was designed to simulate a complex molecular evolution process using a nonparametric population genetic model that generates long-range statistical dependence and heterogeneous rates. Briefly, the simulation is of an RNA molecule with both stabilizing and directional selection on the secondary structure of the molecule. RNA molecules are assumed to mutate both by substitutions and indels. The fixation probability of any potential mutation is computed using a fitness model that is a function of the folded free energy of the mutated RNA. The resulting distribution of evolved molecules display complex statistical characteristics that do not follow the standard parametric models. The simulated dataset displays many of the properties of naturally observed RNA molecules in terms of both morphological variation and optimization difficulty. See Appendix A in the Supplementary Material for more details about RNASim. We subsampled the million-sequence RNASim dataset to create datasets with 10,000, 50,000, 100,000, and 200,000 sequences. For the 10K RNASim dataset we made 10 replicates, but we used only one replicate of these datasets for the larger datasets.
We include three large biological datasets from the comparative ribosomal website (CRW) (Cannone et al., 2002): the 16S.3 dataset (6,323 sequences of average length 1557, spanning three phylogenetic domains), the 16S.T dataset (7,350 sequences of average length 1492, spanning three phylogenetic domains), and the 16S.B.ALL dataset (27,643 sequences of average length 1371.9, spanning the bacteria domain). These datasets have curated reference alignments based on secondary and tertiary structures. Reference trees for the biological datasets were computed using RAxML (Stamatakis, 2006) on the reference alignments and all edges with bootstrap support less than 75% were contracted; using other thresholds produces similar results (see Appendix E.3 in the Supplementary Material). The reference alignments and trees for the simulated datasets are the true alignment and true (model) trees.
4. Results
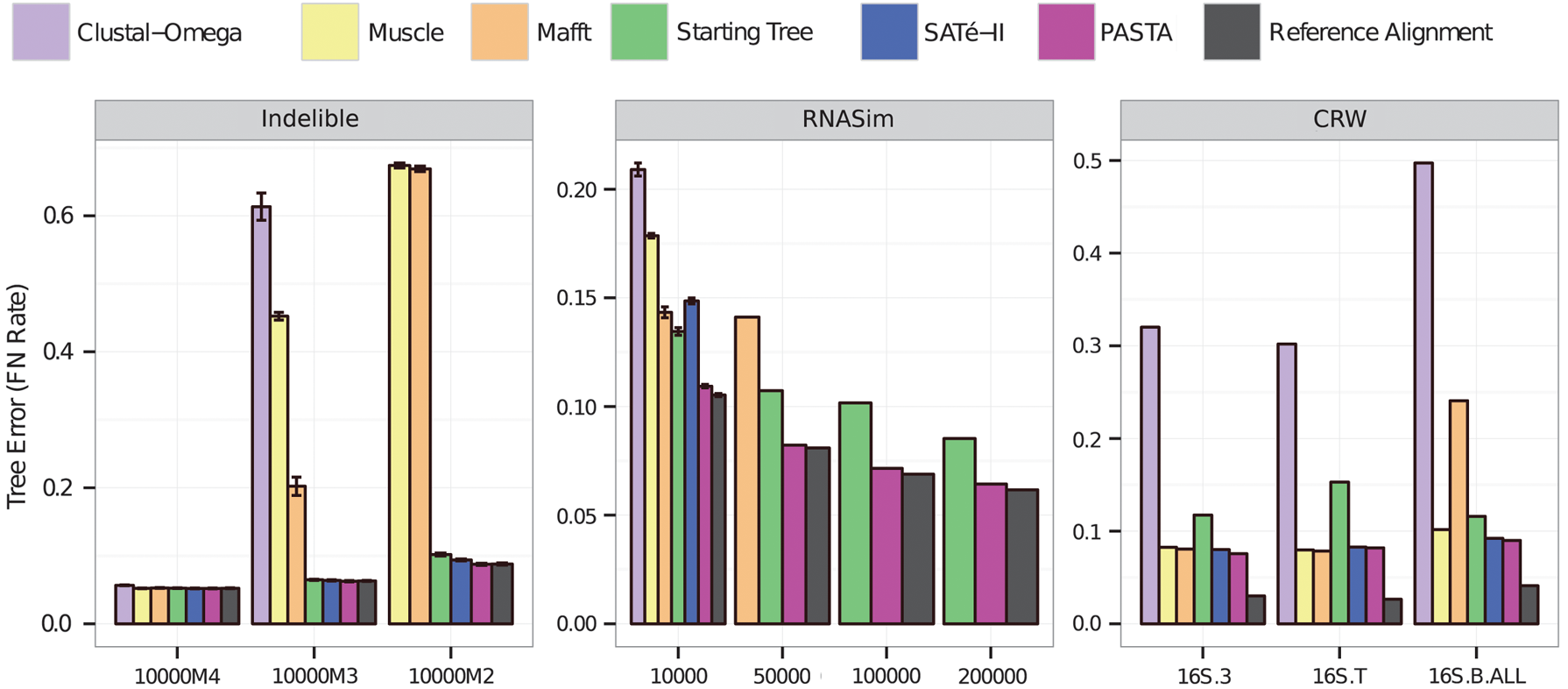
Tree error rates on nucleotide datasets. We show missing branch (also known as false negative or FN) rates for maximum likelihood trees estimated on the reference alignment as well as alignments computed using PASTA and other methods; results not shown indicate failure to complete within 24 hr using 12 cores on the datasets. Error bars show standard error over 10 replicates for all model conditions of the Indelible and the 10,000-sequence RNASim datasets.
Table 1 compares methods with respect to the TC and pairs scores. On the Indelible datasets, PASTA had the most accurate alignments according to both measures of accuracy, and the difference between PASTA and other methods increased as the rate of evolution increased. On the RNASim data, PASTA had by far the most accurate alignments of all methods tested according to TC, and its pairs scores were better than all other methods except for the starting alignment.
We show the number of correctly aligned sites (top) and the average of the SP-score and modeler score (bottom). X indicates that a method failed to run on a particular dataset given the computational constraints. “Initial” corresponds to the alignment approach used to obtain the starting tree of PASTA (HMMER failed to align one sequence in the 16S.T dataset) and Clustal-O stands for Clustal-Omega.
Boldface indicates the best values for each model condition.
On the 16S.T dataset, the starting alignment did not return an alignment with all the sequences, because HMMER considered one of the sequences unalignable; in general, though, the starting alignment had good alignment scores. Of the remaining methods, PASTA had the best pairs scores, and the other methods were substantially less accurate. With respect to TC scores, on 16S.B.ALL and 16S.T, PASTA had the highest accuracy, but on 16S.3, SATé-II had the highest accuracy (followed by Mafft and PASTA).
We show TC (the number of correctly aligned sites, left) and the pairs score (the average of the SP-score and modeler score, right). X indicates that a method failed to run on a particular dataset given the computational constraints. “Initial” corresponds to the alignment approach used to obtain the starting tree of PASTA (HMMER failed to align one sequence in the 16S.T dataset). All values shown are averages over all datasets in each category.
Boldface indicates the best values for each model condition.
PASTA had the best pairs score or was tied for the best pairs score for both HomFam and AA-10 datasets. Mafft had the best TC score for HomFam(17), but PASTA was very close. For HomFam(2), PASTA had the best TC score and Mafft was a close second. On AA-10 datasets, SATé-II had the best TC score and was closely trailed by Mafft and PASTA.
Alignment accuracy and tree error is shown for PASTA with various starting trees, after one iteration (top) and three iterations (bottom) on one replicate of the 10k RNASim dataset.
We also evaluated the impact of changing the alignment subset size (Table 4); these analyses showed that using alignment subsets of only 50 sequences improved the TC score and running time substantially, and only slightly changed the pairs score or tree error score. Although these analyses were performed only for two datasets, they suggest the possibility that improved results might be obtained through smaller alignment subsets.
We report tree error and alignment accuracy on one replicate of the 10K RNASim dataset and also on the 16S.T dataset, using three iterations of PASTA in which we explore the impact of changing the subset size from 200 (the default) to 100 and 50; all other algorithmic parameters use default values. Boldface indicates the best performance on the data.

Alignment running time (hours). Note that PASTA was run for three iterations everywhere, except on the 100,000-sequence RNASim dataset where it was run for two iterations, and on the 200,000-sequence RNASim dataset where it was run for one iteration. Mafft was run in default mode, except for the 100,000-sequences where PartTree was used.
Figure 4a presents a detailed running time comparison of PASTA and SATé-II on two specific model conditions of RNASim dataset. Note that merging subset alignments (and the last pairwise merge, shown in the dotted area) was the majority of the time used by SATé-II to analyze the 50K RNASim dataset, but a very small fraction of the time used by PASTA. PASTA uses transitivity for all but the initial pairwise mergers, and therefore scales well with increased dataset size, as shown in Figure 4b (the sub-linear scaling is due to a better use of parallelism with increased number of sequences). Finally, Figure 4c shows that PASTA is highly parallelizable and has a much better speed-up with increasing number of threads than SATé does.
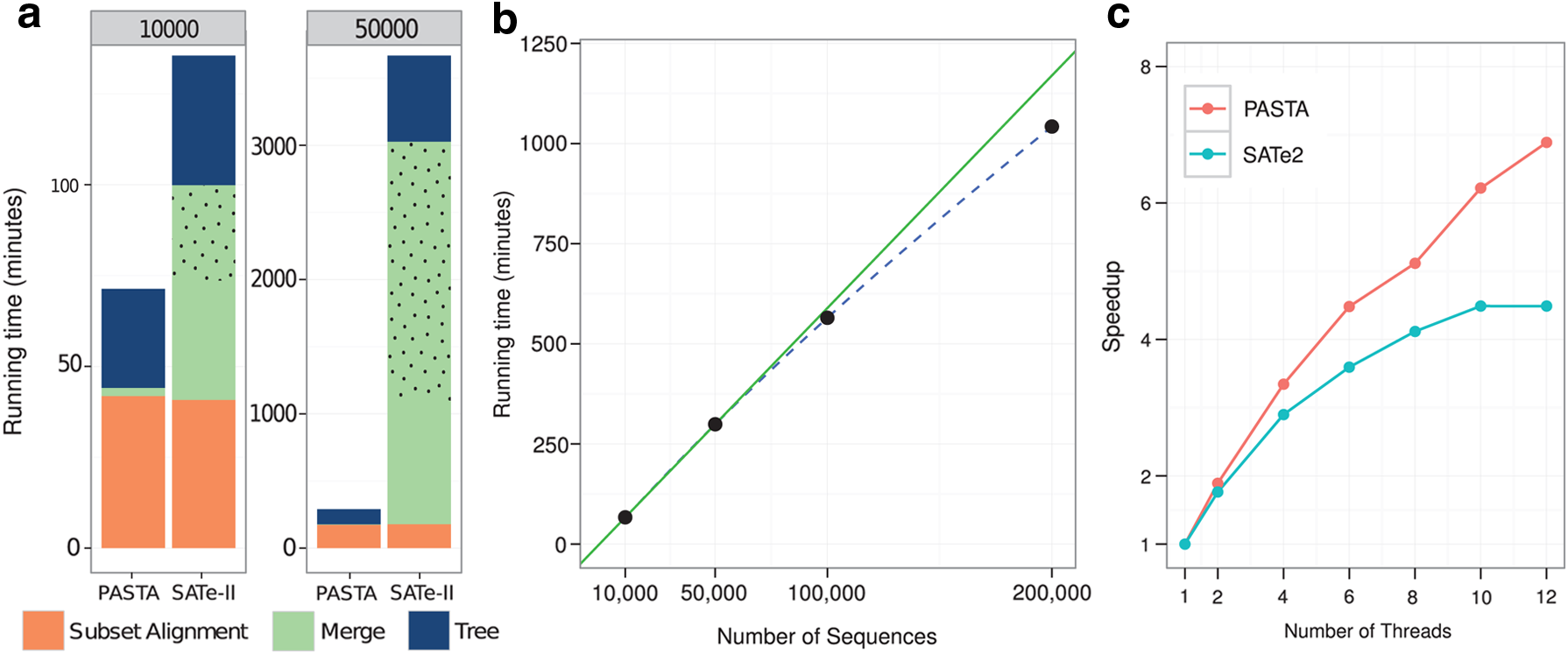
Running time comparison of PASTA and SATé-II.
5. Summary
The key algorithmic contribution in PASTA is the use of transitivity to align sequences on a guide tree, which addresses computational limitations in SATé and also improves the alignment of very distantly related sequences and remote homology detection. PASTA is fast and scales well with the number of processors, so that datasets with even 200,000 sequences can be analyzed in less than a day with a small number of processors. Thus, highly accurate alignment and phylogeny estimation is possible, even on hundreds of thousands of sequences, without supercomputers. (PASTA software is publicly available in open source form online).
Footnotes
Acknowledgments
This research was supported in part by National Science Foundation (NSF) grant DBI 0733029 to T.W., by the Howard Hughes Medical Institute (HHMI) International Predoctoral Fellowship to S.M., and by a subgrant from the University of Alberta to T.W., made possible through a donation from Musea Ventures, which is held by Professor Gane Ka-Shu Wong. This work was also supported by a generous allocation from TACC and by the iPlant Collaborative, NSF grant DBI-1265383. L.W. and S.G. were supported by NSF grant 0331453; L.W. was supported in part by the National Institutes of Health (NIH) through grants T32-HG000046, NIA U24-AG041689, and R01-GM099962. J.K.'s participation in this project was funded, in part, by a Health Research Formula Fund from the Pennsylvania Department of Health, which disclaims responsibility for any analyses, interpretations, or conclusions. The authors thank the anonymous reviewers for their helpful comments.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.