
Abstract
Abstract
The human genome is diploid, which requires assigning heterozygous single nucleotide polymorphisms (SNPs) to the two copies of the genome. The resulting haplotypes, lists of SNPs belonging to each copy, are crucial for downstream analyses in population genetics. Currently, statistical approaches, which are oblivious to direct read information, constitute the state-of-the-art. Haplotype assembly, which addresses phasing directly from sequencing reads, suffers from the fact that sequencing reads of the current generation are too short to serve the purposes of genome-wide phasing
.
While future-technology sequencing reads will contain sufficient amounts of SNPs per read for phasing, they are also likely to suffer from higher sequencing error rates. Currently, no haplotype assembly approaches exist that allow for taking both increasing read length and sequencing error information into account
.
Here, we suggest Whats Hap , the first approach that yields provably optimal solutions to the weighted minimum error correction problem in runtime linear in the number of SNPs. Whats Hap is a fixed parameter tractable (FPT) approach with coverage as the parameter. We demonstrate that Whats Hap can handle datasets of coverage up to 20×, and that 15× are generally enough for reliably phasing long reads, even at significantly elevated sequencing error rates. We also find that the switch and flip error rates of the haplotypes we output are favorable when comparing them with state-of-the-art statistical phasers
.
1. Introduction
T
There are two major approaches to phasing variants. The first class of approaches relies on genotypes as input, which are lists of SNP alleles, together with their zygosity status. While homozygous alleles show on both chromosomal copies, and obviously apply for both haplotypes, heterozygous alleles show on only one of the copies and have to be partitioned into two groups. For m heterozygous SNP positions there are thus 2 m many possible haplotypes. The corresponding approaches are usually statistical in nature, and they integrate existing reference panels. The underlying assumption is that the haplotypes to be computed are a mosaic of reference haplotype blocks that arises from recombination during meiosis. The output is the statistically most likely mosaic, given the observed genotypes. Most prevalent approaches are based on latent variable modeling (Howie et al., 2009; Li et al., 2010; Scheet and Stephens, 2006). Other approaches use Markov chain Monte Carlo techniques (Menelaou and Marchini, 2013).
The other class of approaches uses sequencing read data directly. Such approaches virtually assemble reads from identical chromosomal copies and are referred to as haplotype assembly approaches. Following the parsimony principle, the goal is to compute two haplotypes to which one can assign all reads with the least amount of sequencing errors to be corrected and/or erroneous reads to be removed. Among such formulations, the minimum error correction (MEC) problem has gained most of the recent attention. The MEC problem, which we will formally define in section 2, consists in finding the minimum number of corrections to be made to the sequenced nucleotides in order to arrange the reads into two haplotypes without conflicts. A major advantage of MEC is that it can be easily adapted to a weighted version (wMEC) in order to deal with phred-scaled error probabilities. Such phred-based error schemes are vital, in particular for processing long reads generated by future technologies, as these are prone to elevated sequencing error rates. An optimal solution for the wMEC problem then translates to a maximum likelihood scenario relative to the errors to be corrected.
In tera-scale sequencing projects (e.g., Boomsma et al., 2013; The 1000 Genomes Project Consortium, 2010), ever increasing read length and decreasing sequencing cost make it clearly desirable to phase directly from read data. However, statistical approaches are still the methodology of choice because (i) most NGS reads are still too short to bridge so-called variant deserts. Successful read-based phasing, however, requires that all pairs of neighboring heterozygous SNP alleles are covered; and (ii) the MEC problem is
Most advanced existing algorithmic solutions to MEC (Chen et al., 2013; He et al., 2010) ironically often benefit precisely from variant deserts, because these allow for decomposing a problem instance into independent parts. A major motivation behind read-based approaches, however, is to handle long reads that cover as many variants as possible, thereby bridging all variant deserts. Hence, the current perception of haplotype assembly is often that it underlies theoretical limitations that are too hard to overcome.
Here, we present W
2. The Weighted Minimum Error Correction Problem
The input to the MEC problem is a matrix
A haplotype can formally be defined as a string of length m consisting of 0's and 1's. If h is a haplotype, then the i-th row of
The goal of MEC is to make
The MEC problem, which is also called minimum letter flip, was introduced by Lippert et al. (2002). Cilibrasi et al. (2005) showed that this problem is NP-hard even if each fragment is “gapless,” that is, if it consists of a consecutive sequence of 0's and 1's with holes to the left and to the right. Panconesi and Sozio (2004) were the first to propose a practical heuristic for solving MEC. An exact branch and bound algorithm and a heuristic genetic algorithm were presented by Wang et al. (2005). Levy et al. (2007) designed a greedy heuristic to assemble the haplotype of the genome of J. Craig Venter. Bansal et al. (2008) developed an MCMC method to sample a set of likely haplotypes. In a follow-up, some of the authors proposed a much faster MAX-CUT-based heuristic algorithm called H
The weighted variant of MEC was first suggested by Greenberg et al. (2004). Zhao et al. (2005) proposed a heuristic for a special case of wMEC and presented experiments showing that wMEC is more accurate than MEC. In the next section, we presented a dynamic programming algorithm for wMEC, which is similar in spirit to the approach of Deng et al. (2013), but extends it to the weighted case and broadens its applicability using techniques from algorithm engineering.
3. A Dynamic Programming Algorithm for Wmec
We now present the W
Consider the input matrix
Our dynamic programming (DP) formulation is based on the observation that, for each column, only active fragments need to be considered; a fragment i is said to be active in every column j that lies in between its leftmost nonhole entry and its rightmost nonhole entry. Thus, paired-end reads remain active in the “internal segment” between the two reads. Let F(j) be the set of fragments that are active at SNP position j and let F be the set of all fragments. The aim is to find a bipartition (R*, S*) of F such that the changes in R* and S* to make
Proceeding columnwise from SNP position 1 to m, our approach computes a DP table column C(j,·) for each
The basic idea of our dynamic program is as follows: for every bipartition B=(R, S) of F(j), entry C(j, B) gives the minimum cost of a bipartition of all fragments F that renders positions
To compute the contribution Δ C (j, (R, S)) of column j to the cost C(j, (R, S)) of bipartition (R, S), we define the following quantities.
Hence, given a bipartition (R, S) of F(j), the minimum cost to make position j conflict-free is
Notice that, under the all heterozygous assumption, where one wants to enforce all SNPs to be heterozygous, the equation becomes
In both cases, we only need the four values W0(j, R), W1(j, R), W0(j, S), and W1(j,S) to compute Δ C (j, (R, S)). We now proceed to state in detail our DP formulation.
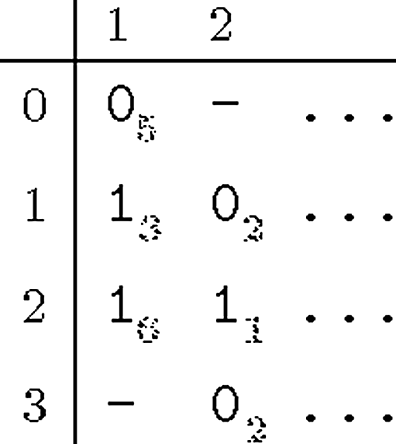
Note that we need consider only half of the 2|F(1)| bipartitions, because C(j, (R, S))=C(j, (S, R)) for every bipartition B=(R, S) and every SNP position j.
The first term accounts for the cost of the current SNP position, while the second term accounts for costs incurred at previous SNP positions. The minimum selects the best score with respect to the first j positions over all partitions that extend (R, S).
for all
For an efficient computation of Δ
C
(j, Bj(k)), we enumerate all bipartitions
To efficiently implement the DP recursion, one can compute an intermediate projection column as follows. For all
Table
The algorithm has a runtime of O(2k−1m), where k is the maximum value of cov(·), and m is the number of SNP positions. Note that the runtime is independent of read length.
An optimal bipartition can be obtained by backtracking. To do this efficiently, we store tables
Using these auxiliary tables, the sets of fragments that are assigned to each allele can be reconstructed in O(km) time. To backtrace an optimal bipartition, we need to store the rightmost DP column C(m,·) and the backtracking tables
Backtracking gives us optimal fragment bipartitions
4. Experimental Results
The focus of this article is on very long reads and their error characteristics. Since such data sets are not available today, we performed a simulation study where we simulated long, future-generation reads, and also current-generation reads, to compare current and future technologies with respect to read-based phasing. We used all variants—that is, SNPs, deletions, insertions, and inversions—reported by Levy et al. (2007) to be present in J. Craig Venter's genome. These variants were introduced into the reference genome (hg18) to create a reconstructed diploid human genome with fully known variants and phasings. Using the read simulator SimSeq (Earl et al., 2011), we simulated current-generation sequencing reads using HiSeq and MiSeq error profiles, to generate a 2 × 100 bp and a 2 × 250 bp paired-end data set, respectively. The distribution of the internal segment size (i.e., fragment size minus size of read ends) was chosen to be 100 bp and 250 bp, respectively, which reflects current library preparation protocols. Longer reads with 1,000 bp, 5,000 bp, 10,000 bp, and 50,000 bp were simulated with two different uniform error rates of 1 % and 5 %. All data sets were created to have 30× average coverage and were mapped to the human genome using BWA MEM (Li, 2013).
To avoid confounding results by considering positions of wrongly called SNPs, we always used the set of true positions of heterozygous SNPs that were introduced into the genome. We extracted all reads that covered at least two such SNP positions to be used for phasing. Next, we pruned the data sets to target coverages of 5×, 10× and 15× by removing randomly selected reads that violated the coverage constraints until no more such reads exist. The resulting sets of reads were finally formatted into matrix-style input, needed as input for most haplotype assembly approaches. In our case, weights correspond to phred-scaled error probabilities. That is, for example, a weight of X corresponds to probability 10−(X/10) that the corresponding matrix entry (0 or 1) is wrong due to a sequencing error.
4.1. Comparison of WhatsHap to other methods
To our knowledge, no other methods exist that can solve instances of wMEC with very long reads to optimality in practice. However, there is a fairly mature body of research and resulting implementations that solve the unweighted MEC problem, both heuristically and exactly. Here we compare W
We ran all methods on a 12-core machine with Intel Xeon E5-2620 CPUs and 256GB of memory. We compared the runtime of these three methods against ours, for chromosome 1 of J. Craig Venter's genome (Levy et al., 2007) of our simulated reads dataset. Since all other methods are restricted to the unweighted case, we ran W
Table 1 shows the runtimes of the four tools for coverages 5×, 10×, and 15×. Runtimes for the general case, in which haplotypes may also contain homozygous positions, are given in the Supplementary Material (available online at www.liebertpub.com/cmb). We observe that the method by He et al. (2010) cannot solve but one instance within the time limit of 5 hr and the memory limit. The runtimes of both W
A “—” stands for an unsuccessful run, either because it exceeded the time limit of 5 CPU hours, or it exceeded all of the available memory.
4.2. Accuracy of WhatsHap
The accuracy performance is summarized in Figure 2. There, the percentage of chromosome 1 that could not be phased due to missing information (unphasable positions: y-axis) is plotted against the percentage of positions in the haplotypes predicted that are affected by errors. We display these percentage rates for all datasets and all coverages. For the “future-generation” reads, we show results for a sequencing error rate of 1% and note that we provide a detailed phase error analysis also for higher sequencing error rates (5%). A respective plot (analogous to Fig. 2) can be found in the Supplementary Material.

Performance of phasing human chromosome 1 with 68 184 heterozygous SNPs in total using different simulated data sets and different coverages. The unphasable positions percentage (y-axis) gives the fraction of the SNP positions that could not be phased due to not being covered by reads that span more than one SNP position. The x-axis shows the percentage of all SNPs that were not unphasable but wrongly phased by the algorithm, either because of a flip error, a switch error, or due to being reported as ambiguous position by
We call a SNP position uncovered if it is not covered by any read in the data set. We denote a break as two consecutive SNP positions that are not physically bridged by any read in the dataset. Finally, we define the number of unphasable positions to be the number of uncovered SNP positions plus the number of breaks—we are, of course, aware of the fact that breaks do not one-to-one refer to SNP positions, but to positions in between two consecutive SNP positions (which overall amount to the number of SNP positions minus one). We neglect this here for the sake of simplicity—note that breaks and uncovered positions never interfere with one another such that our counting scheme is not ambiguous.
We further distinguish between three classes of errors:
1. Flip errors, which are errors in the predicted haplotypes that can be corrected by flipping an isolated 0 to a 1 or vice versa. As an example, consider the correct haplotype pair to be 000|111, while the prediction is 010|101: the second position is then affected by a flip error. 2. Switch errors are two consecutive SNP positions whose phases have been mistakenly predicted, and which cannot be interpreted as flip errors. For example, if the correct haplotype is 000111|111000 and the predicted haplotype is 000000|111111, then we count one switch error between positions 3 and 4. 3. Ambiguity errors are SNP positions where flipping 0 and 1 does not lead to decreasing the (w)MEC score. One can expect an error for half of these positions, because they are, effectively, phased randomly.
The x-axis displays the sum of all such errors over the overall amount of SNP positions.
Figure 2 shows that, while current-generation sequencing reads (HiSeq and MiSeq) still leave substantial amounts of positions unphasable, long reads dramatically decrease the number of unphasable positions. One further notices that error rates are very favorable (only between 0.1 and 0.25% of positions are affected by phasing errors), when comparing them to error rates that can be achieved by statistical phasers, which usually amount to at least 1%; see, for example, O'Connell et al. (2014) (Table 1 on “unrelated individuals”) for evaluations of such methods. It is further noteworthy that higher sequencing error rates do not lead to drastic effects in these statistics, for the weighted case (see a detailed analysis in Benefits of Weights below). Furthermore, we find that the importance of high coverage is rather limited. Phasing error rates do not increase drastically after having reached a coverage of 10–15× such that one can conclude that such coverage rates are sufficient for next- and future-generation sequencing read–based haplotype assembly.
4.3. Benefits of weights
W
For each of these error categories, numbers for solving the unweighted (uw) and weighted (w) case are given.
5. Conclusions and Further Work
The increased length of future-generation sequencing reads comes with obvious advantages in haplotype assembly, because it allows for linking ever more SNP positions based on read information alone. However, future-generation reads may also be subject to elevated sequencing error rates, which can result in elevated phasing error rates. Here, we have presented W
While our approach handles datasets with possibly long reads, it can only deal with limited coverage. Although W
In the literature there are several graph representations of haplotype data (the fragment conflict graph defined in Lancia et al., 2001, and many of its variants), and consequently the optimization problems we have mentioned are seen there as finding the minimum number of graph editing operations that make the graph bipartite. In particular, for the conflict graph variant used in Fouilhoux and Mahjoub (2012), the MEC problem turns out to be equivalent to finding the maximum induced bipartite subgraph (MIBS). It follows that our dynamic programming approach for MEC can be generalized to an FPT approach for MIBS where the parameter is the pathwidth of the graph.
In this work we have concentrated on assembling SNP haplotypes from reads of a sequenced genome. As a next step we will integrate predictions from statistical phasers into our approach. In some sense, the super-read obtained from a slice, mentioned above, can be viewed as a reference haplotype from a reference panel for an existing population. Hence, reference haplotypes can be seamlessly integrated into this merging step (iii) for a hybrid approach. Hybrid methods are the future of sequencing data analysis, and the field is already moving quickly in this direction (Delaneau et al., 2013; He et al., 2013; He and Eskin, 2013; Selvaraj et al., 2013; Yang et al., 2013; Zhang, 2013).
In addition, haplotyping mostly refers to only SNPs for historical reasons (The International HapMap Consortium, 2007, 2010). To fully characterize an individual genome, however, haplotyping must produce exhaustive lists of both SNPs and non-SNPs, that is, larger variants. This has become an essential ingredient of many human whole-genome projects (Boomsma et al., 2013; The 1000 Genomes Project Consortium, 2010). In this article we focus on SNP variants, and we identify the integration of non–SNP variants as a challenging future research direction.
Lastly, we used prior knowledge of the true SNP positions in the genome in our simulation study. But since our method only scales linearly in the number of SNP positions, one could conceivably also use the full raw read input to produce a “de novo” haplotype. Since SNPs comprise roughly 5% of positions, and the runtime of our method is on the order of 10 min on average (for sufficient 15× coverage), such a de novo haplotype could be generated in about 3 hours. The heterozygous sites of this constructed haplotype then correspond to the SNP positions. It hence follows that this tool could be used for SNP discovery, and perhaps for larger variants as well.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Acknowledgments
Murray Patterson was funded by a Marie Curie ABCDE Fellowship of ERCIM. Leo van Iersel was funded by a Veni, and Alexander Schönhuth was funded by a Vidi grant of the Netherlands Organisation for Scientific Research (NWO).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.