
Abstract
Abstract
Compressed sensing (CS) is a sparse signal sampling methodology for efficiently acquiring and reconstructing a signal from relatively few measurements. Recent work shows that CS is well-suited to be applied to problems in genomics, including probe design in microarrays, RNA interference (RNAi), and taxonomic assignment in metagenomics. The principle of using different CS recovery methods in these applications has thus been established, but a comprehensive study of using a wide range of CS methods has not been done. For each of these applications, we apply three hitherto unused CS methods, namely, l1-magic, CoSaMP, and l1-homotopy, in conjunction with CS measurement matrices such as randomly generated CS m matrix, Hamming matrix, and projective geometry-based matrix. We find that, in RNAi, the l1-magic (the standard package for l1 minimization) and l1-homotopy methods show significant reduction in reconstruction error compared to the baseline. In metagenomics, we find that l1-homotopy as well as CoSaMP estimate concentration with significantly reduced time when compared to the GPSR and WGSQuikr methods.
1. Introduction
C
There are numerous examples of CS being applied to the field of genomics. CS has been applied to genome-wide association studies (GWAS) as well as genomic selection (Vattikuti et al., 2014). The aim of GWAS is to identify trait-associated loci, which are DNA segments containing genes associated with some phenotype, whereas genomic selection attempts to predict the phenotypic values for new individuals based on all possible short segments of DNA containing genes. CS addresses a problem common to both endeavors—that the number of genotyped markers often greatly exceeds the sample size. CS-based methods have also been used for identification of rare alleles and their carriers where short DNA segments containing genes for a large set of individuals have to be sequenced, as opposed to sequencing large segments of DNA in a single individual (Shental et al., 2010).
The most detailed of CS applications in genomics has been in the field of DNA microarrays (Dai et al., 2009; Yok and Rosen, 2008). A DNA microarray is an array of large numbers of DNA sequences spotted onto a solid surface. It is used to measure the expression levels of large numbers of genes simultaneously. Each DNA spot contains a minute concentration of a specific DNA sequence, called a “probe.” These probes can be short sections of a gene or some other DNA element, and are used to hybridize another DNA sample called the “target” (Trevino et al., 2007). Probe–target hybridization is detected and quantified by detection of spot intensity to determine relative abundance of DNA sequences in the target. The implementation cost and speed of microarray data processing depends on the number of probes. Microarrays can be miniaturized by reducing the number of probes to be much less than the number of targets since a large number of probes remains unused or inactive, making the nature of a microarray read sparse. A microarray designed with CS principles of having reduced number of probes is called a compressed sensing microarray or CSM (Dai et al., 2009).
Another area of genomics where CS has been applied is RNA interference (RNAi). RNAi is the process by which certain RNAs, known as small interfering RNAs (siRNAs), inhibit gene expression by causing the degradation of specific mRNAs (Mohr and Perrimon, 2012). Functionally, one siRNA knocks down only one mRNA target. However, a conceptual model called compressed sensing RNAi (csRNAi) has been developed, which employs a unique combination of a group of siRNAs to knockdown a much larger number of target mRNAs (Tan et al., 2012). This involves a many-to-many correspondence between siRNAs and their target mRNAs, with the number of siRNAs being lesser than the mRNA targets (Tan et al., 2012).
CS has also been applied to metagenomics that involves genomic analysis of microorganisms by direct extraction and cloning of DNA from a mixed sample of such microorganisms (Handelsman, 2004). Shotgun sequencing allows for sampling all genes for all microorganisms in a particular metagenomic sample. However, the large number of relatively short reads output from such technologies has necessitated fast and accurate short-read classifiers. Traditional methods classify samples in a read-by-read manner, leading to execution times ranging from hours to days (Koslicki et al., 2013). However, CS techniques have been successfully applied resulting in faster processing and increased accuracy (Amir and Or, 2011; Koslicki et al., 2014).
In many of these genomic applications, the principle of using CS to solve the problem has been previously established. Thus, CSMs help by reducing the number of probes to be much smaller than the number of targets; CS matrices based on the affinity between siRNAs and target sequences in a many-to-many correspondence have been used for successful signal recovery, and CS methods can compute taxonomic assignments with good speed and accuracy. However, a comprehensive study of using a wider range of CS methods and matrices in these applications has not been done. We propose to use hitherto unused CS recovery methods in conjunction with a variety of matrices in these genomic applications.
2. Background
2.1. A Note on norms
An important mathematical concept to understand is a norm. Mathematically, a norm is the total size or length of all vectors in a vector space or matrices. Formally, l
n
-norm of x is defined in Equation 1:
The most popular of all norms is the l2-norm. It is used in almost every field of engineering and science. Following the basic definition, l2-norm is defined in Equation 2:
l2-norm is well known as a Euclidean norm, which is used as a standard quantity for measuring a vector difference. For a vector difference, it is known as a Euclidean distance given in Equation 3:
Error is measured in terms of mean-squared error (MSE) measurement, which is used to compute a similarity or a correlation between two signals. MSE is given in Equation 4:
2.2. CS basics and recovery methods
CS has been very active in the community of signal processing theory and applications in the last decade. Although started as a framework to acquire sparse or compressible signals with linear measurements at a rate much smaller than the Shannon–Nyquist criteria, the powerful ramifications of sparseness and the associate processing led to diverse penetration of CS.
In CS, we can recover the sparse vector x (say of length N and sparsity s) from M with s < M < N measurements through the operation
if the measurement matrix Φ satisfies certain conditions based on one of these desirable properties: null-space property (NSP), restricted isometry property (RIP), or coherence (Davenport, 2013). The recovery of x can be carried out using
This is formulated as a linear program (LP) in the real case and a second-order cone program (SOCP) in the complex case, often referred to as basis pursuit (Fornasier and Rauhut, 2011). Both LP and SOCP can be solved with interior point methods as in l1-magic package (Candès and Romberg, 2005).
However, the computational complexity of these general-purpose algorithms and the associated standard software is often too high for many real-world, large-scale applications (Yang et al., 2013). Toward addressing this complexity, different algorithms, such as matching pursuit (MP) (Mallat and Zhang, 1993), orthogonal matching pursuit (OMP) (Tropp and Gilbert, 2007), compressive sampling matching pursuit (CoSaMP) (Needell and Tropp, 2008), and subspace pursuit (SP) (Dai et al., 2009), have been proposed. In order to account for the measurement noise in realistic applications, it is necessary to work with the relaxed constraints. These include
In homotopy, the following optimization is considered, which is an
The method traces the parameterized solution
Apart from the aforementioned reconstruction algorithms, there are other CS recovery methods applied in the field of genomics. For instance, in gradient projection for sparse reconstruction or GPSR (Mário et al., 2007), the convex relaxation problem of Equation 7 is first turned into a bound-constrained quadratic program by separating the solution into its positive and negative parts. This is then solved by iterative gradient projection algorithms, among others. Another well-studied decoding strategy in microarrays is through belief propagation using the sum-product algorithm over factor graphs (Yedidia and Freeman, 2003). Belief propagation facilitates evaluation of marginal probabilities (Sarvotham et al., 2006). The mapping of the CS microarray decoding to belief propagation over factor graph has been explained in detail (Dai et al., 2009). In microarrays, as the target concentration increases, saturation effect becomes dominant, causing nonlinearity in the measurement intensities representing the concentration levels of targets. Belief propagation strategy allows for elegant handling of these nonlinearities (Sarvotham et al., 2006; Dai et al., 2009).
Hale's open codes (Hale et al., 2007) related to fixed-point continuation (FPC) algorithm have also been used to solve large-scale l1-regularized least-squares problems with reduced time. Here, Equation 8 is minimized using fixed-point iterations paired with a continuation strategy for λ to reduce the number of iterations. This is based on two ideas, namely, forward–backward splitting and continuation (Hale et al., 2007). It is to be noted that, in Equation 8, M is any m × m positive definite matrix, or the empty matrix and the norm with respect M is defined through the quadratic form.
WGSQuikr refers to Whole Genome Shotgun Quikr, where Quikr stands for QUadratic, K-mer based, Iterative, and Reconstruction (Koslicki et al., 2013, 2014) developed for metagenomics. Here, the taxonomic assignments of an entire metagenome are computed, instead of computing the assignments in a read-by-read fashion. It is a nonnegative basis pursuit denoising algorithm that can be reduced to a nonnegative least squares problem. The sensing matrix is obtained by taking frequency of occurrence of k-mers in a known microorganism genome database, where k-mers refer to all the possible subsequences of length k from a read obtained through sequencing. Given a microorganism community represented in a database as a sparse vector x with small microorganism concentration levels, the sparsity-promoting minimizations involving the l1-norm are used to recover the microorganism concentrations.
2.3. Measurement matrices
There are different “measurement” matrices that have been used in the various genomics-related CS applications. These include traditional randomly matrices (Yok and Rosen, 2008; Tan et al., 2012), Hamming matrices (Dai et al., 2009), and low-density parity check (LDPC) matrices (Sheikh et al., 2007). As a novelty, we have considered projective geometry (PG)-based matrices and compared these against the performance of the aforementioned matrices (Dai et al., 2009; Yok and Rosen, 2008; Tan et al., 2012), to evaluate their performance when different recovery methods are used.
3. Methods
MATLAB (2012) has been used for implementation of the recovery methods and sensing matrices. Implementation of the different recovery methods have been used, such as l1-magic (Candès and Romberg, 2005) and belief propagation (Sarvotham et al., 2006).
3.1. Projective geometry matrix
LDPC codes have established themselves as useful codes for different applications in the last decade. They are characterized by sparse parity check matrices and there are different ways to design them (see Lin and Costello, 2004; Johnson, 2006 and the references therein). In this work, we have considered cyclic codes based on finite PG of dimension two (Lin and Costello, 2004; Attili, 2006). The latter plane PG is characterized and represented by
In the usual design of PG codes, the parity check matrix is an n × n square matrix obtained from the incidence relationship between the lines and the points. For instance, a
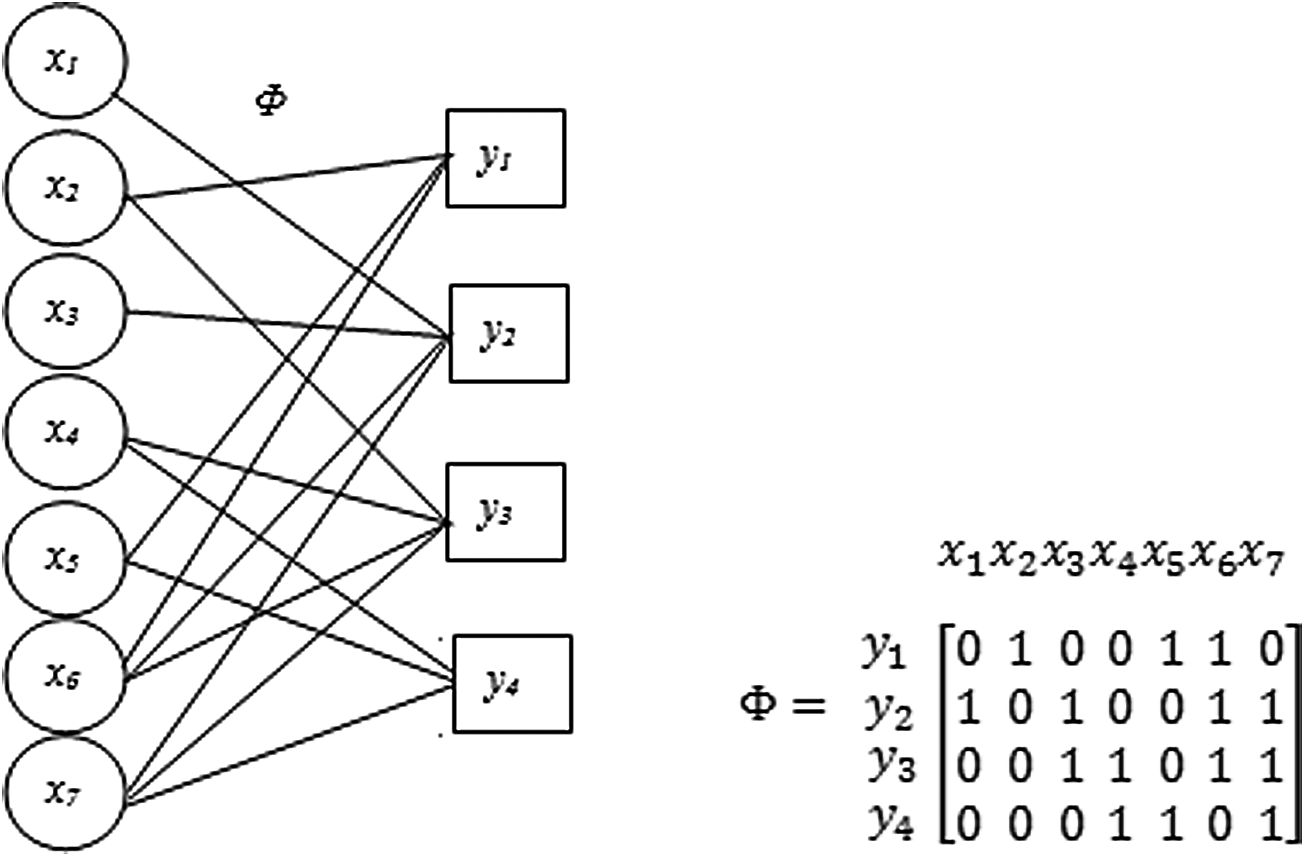
Factor graph in compressive sensing using belief propagation. Factor graph showing the relation between variable nodes x's and y's constraint nodes for 4 × 7 matrix.
3.2. Compressed sensing microarray
The design starts with selecting N targets of desired length and a reference binary CS matrix Φ of size M × N. The goal is to obtain M probes where the hybridization affinity between any ith probe and jth target is approximately equal to the value of
These target sequences are originally from the COG database (Tatusov et al., 2003). We chose the target sequence length as 55 bp and there are a total of 7 targets. Four probes of length 70 are obtained using algorithms 1 and 2. The alignment length threshold was set to 20, indicating good binding between probe and target sequences (Dai et al., 2009; Yok and Rosen, 2008). A 4 × 7 matrix was used as the binary CS matrix. We used MATLAB function swalign with default gap open and extension penalty of 8, and percent identity threshold of 0.67 to exactly implement the binary CS matrix of the baseline (Dai et al., 2009). In algorithm 2, probe sequences of 55 bp length were generated for comparison purpose, as opposed to selecting 26 bp probes from entire genomic sequence of groups. For a row satisfying the desired binding pattern, its corresponding probe was selected. Using the COG datasets of seven targets and four probes generated from probe design algorithms, the matrix and recovery method combination were tested for effectiveness (Dai et al., 2009).
Furthermore, it has been reported that the signal acquisition model becomes nonlinear due to nonlinearity in measurement intensities for a single spot-single target situation (Sheikh et al., 2007). Steps to solve nonlinearity were taken from Dai et al. (2009).
3.3. Compressed sensing RNAi
The siRNA probe sequences were selected from an EST dataset containing 600 elements. This dataset is part of the larger UniGene cDNA library NCI_CGAP_DHO that contains a total of 4647 ESTs related to VS-8 cell line from lung metastatic chondrosarcoma (Tan et al., 2012). Using an artificial neural network implementation called BIOPRED (Huesken et al., 2005) and the 8 characteristics for siRNA design (Reynolds et al., 2004), a 48 × 146 network corresponding to affinity between siRNA and ESTs was identified (Tan et al., 2012).
Due to nonavailability of the 48 × 146 PG matrix, we chose a matrix that was closest to test all the 146 target genes. The closest available matrix was a 28 × 73 one. All 146 targets could be detected by 48 potential siRNA probes by dividing them into 2 sets of 73 sequences each. However, out of 48 available potential probes, 28 are chosen that approximately satisfy the binding pattern of the matrix (Tan et al., 2012). This PG matrix of 28 × 73 is used in conjunction with recovery methods and compared.
3.4. Metagenomics
The recovery methods are separately tested on sample dataset of bacterial genomes to estimate concentration (Koslicki et al., 2014). In a database of 2,483 unique sequences. These were retrieved, along with their taxonomic information. The sensing matrix Φ is the frequency of k-mers (for a fixed k = 7). The matrix is not positive definite; hence, recovery methods are applied separately on the dataset. The concentrations of the bacteria in the sample are reconstructed by solving a system of linear equations. Thus, the problem is reduced to finding vector x that gives the bacterial concentration by solving the general CS equation
The entire methodology followed in this work is captured in Figure 2. For this work, we have used a laptop having an Intel Core i5-2430M processor and 6 GB RAM, and running 64-bit Windows 7 Operating System.
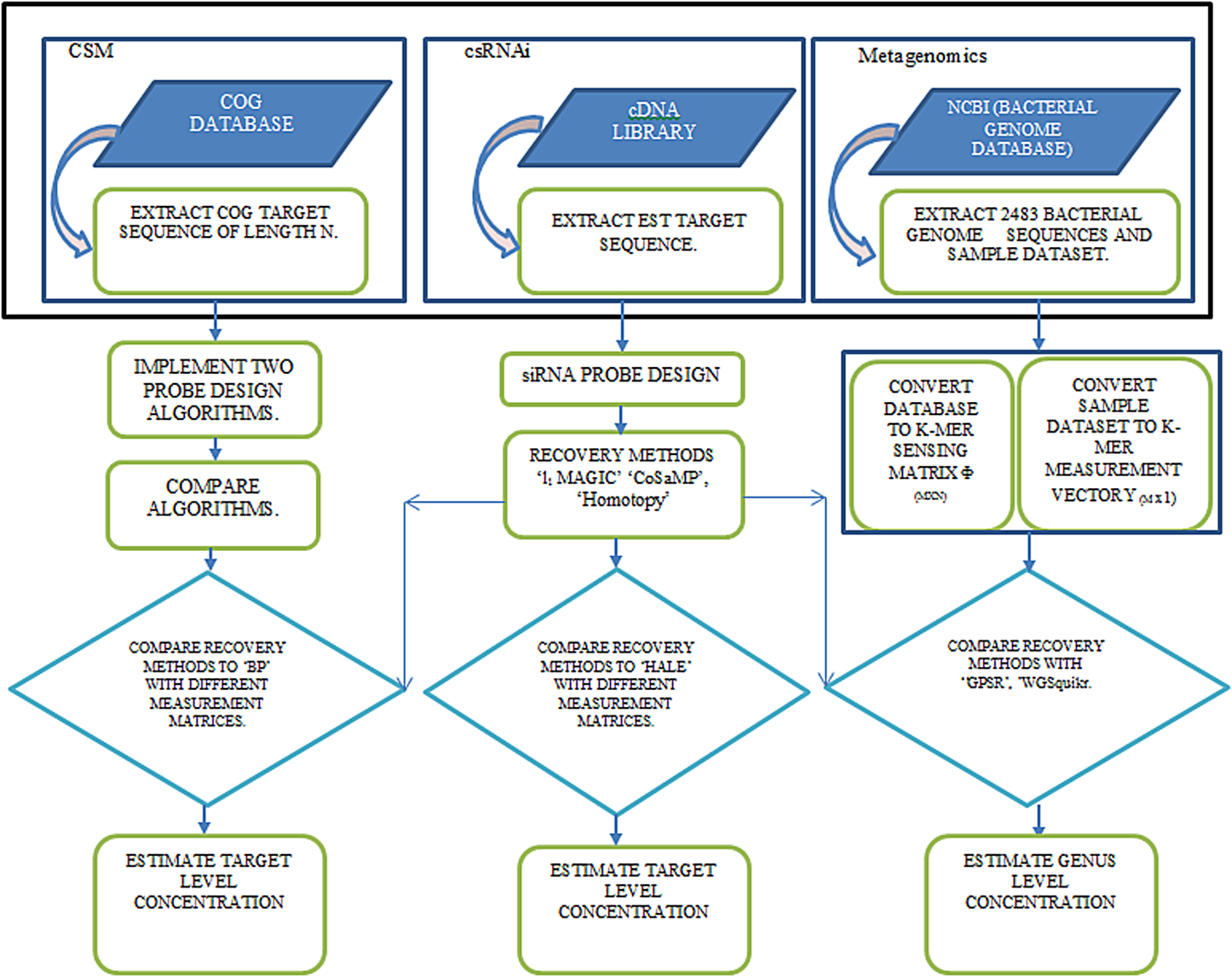
Block diagram of the process. The block diagram represents the procedure followed for compressed sensing microarray (CSM) probe design, compressed sensing RNAi (csRNAi) probe selection, and for metagenomics application, with detailed description given in the Methods section.
4. Results
4.1. Compressed sensing microarrays
The PG matrix of size 4 × 7 was tested on COG dataset of 7 targets and 4 probes generated using probe design algorithms 1 and 2. Target concentration sparse vector x of length 7 × 1 is randomly generated that is considered as original concentration; reconstruction error is calculated between the original concentration x and reconstructed xr concentration from recovery methods. PG gave the least reconstruction error when compared to Hamming matrix and random CS matrix.
As shown in Figure 3, the three recovery methods, l1-magic, l1-homotopy, and CoSaMP, perform significantly better when compared with belief propagation with respect to reconstruction error. Additionally, belief propagation takes more time compared to other methods, as shown in Figure 4. Time taken by a recovery method when applied on different matrices remains same, with negligible variation. Table 1 summarizes the comparisons of recovery methods with matrices.
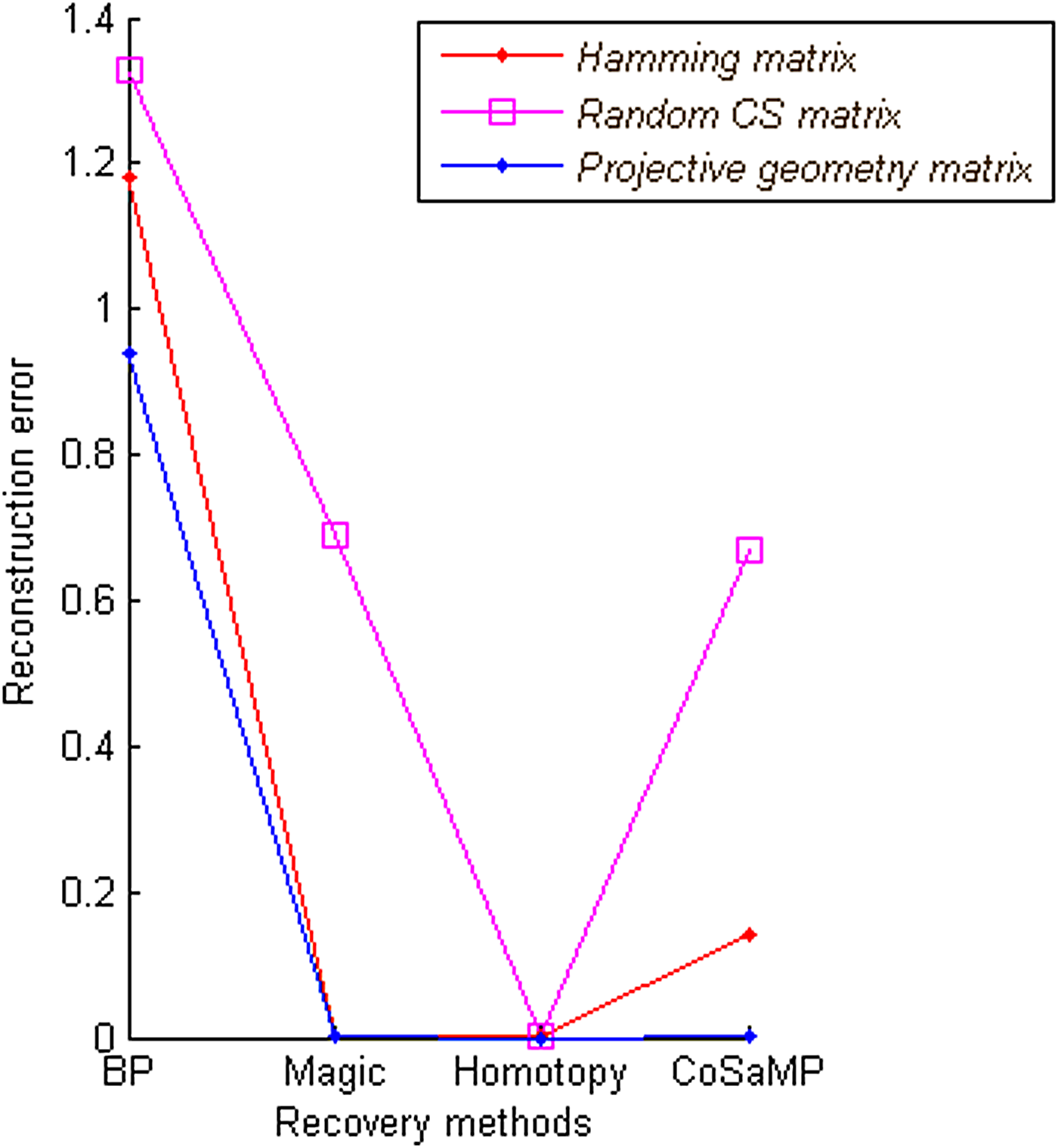
Comparison of recovery methods with respect to reconstruction error. The recovery methods with projective geometry (PG) matrix are compared to baseline basis pursuit (BP) recovery methods, and matrices Hamming and random compressed sensing (CS) on small-scale COG dataset.
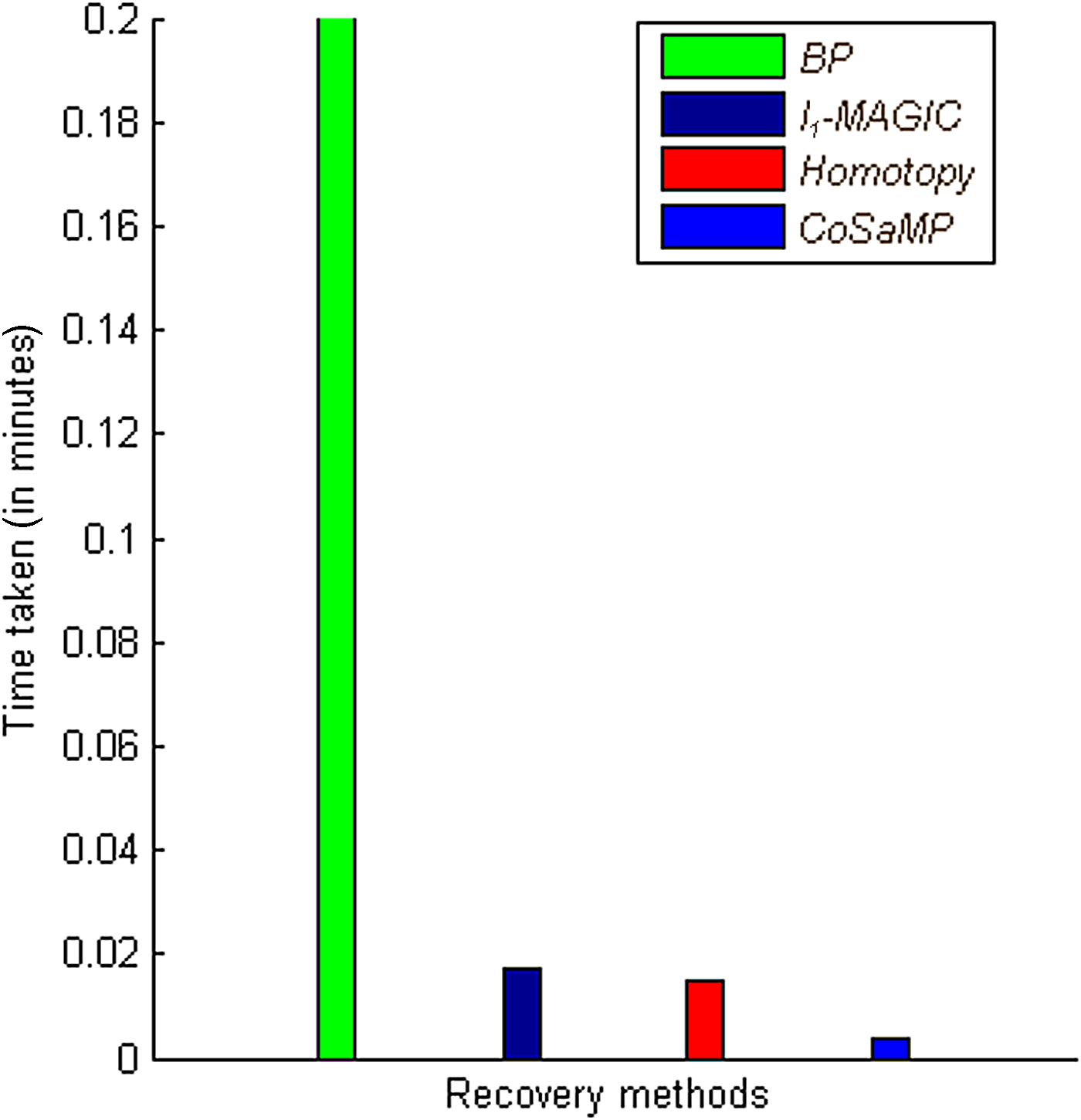
Comparison of recovery methods with respect to time. Computational time taken by recovery methods to recover a target concentration for small scale COG datasets is compared.
Summary of the comparison of recovery methods in conjunction with different matrices of size 4 × 7 that are used as binding patterns in CSM probe design. CS, compressed sensing; CSM, compressed sensing microarray; PG, projective geometry.
4.2. Compressed sensing RNAi
PG matrix of size 28 × 73 was tested on EST dataset of 73 targets and 28 siRNA probes was compared to the baseline matrix random CS matrix. The PG matrix gives the significantly lesser reconstruction error. Using this PG matrix, recovery methods were tested. As shown in Figure 5, l1-magic (the standard package for l1 minimization) performs better when compared with baseline Hale's open codes with respect to the reconstruction error. Time taken by recovery methods when applied on different matrices remains same, with negligible variation (Fig. 6). Table 2 summarizes the RNAi results.

Comparison of recovery methods with respect to reconstruction error (RNAi). The recovery methods with PG matrix are compared to the baseline Hale's open codes recovery method and random CS matrix on EST dataset.
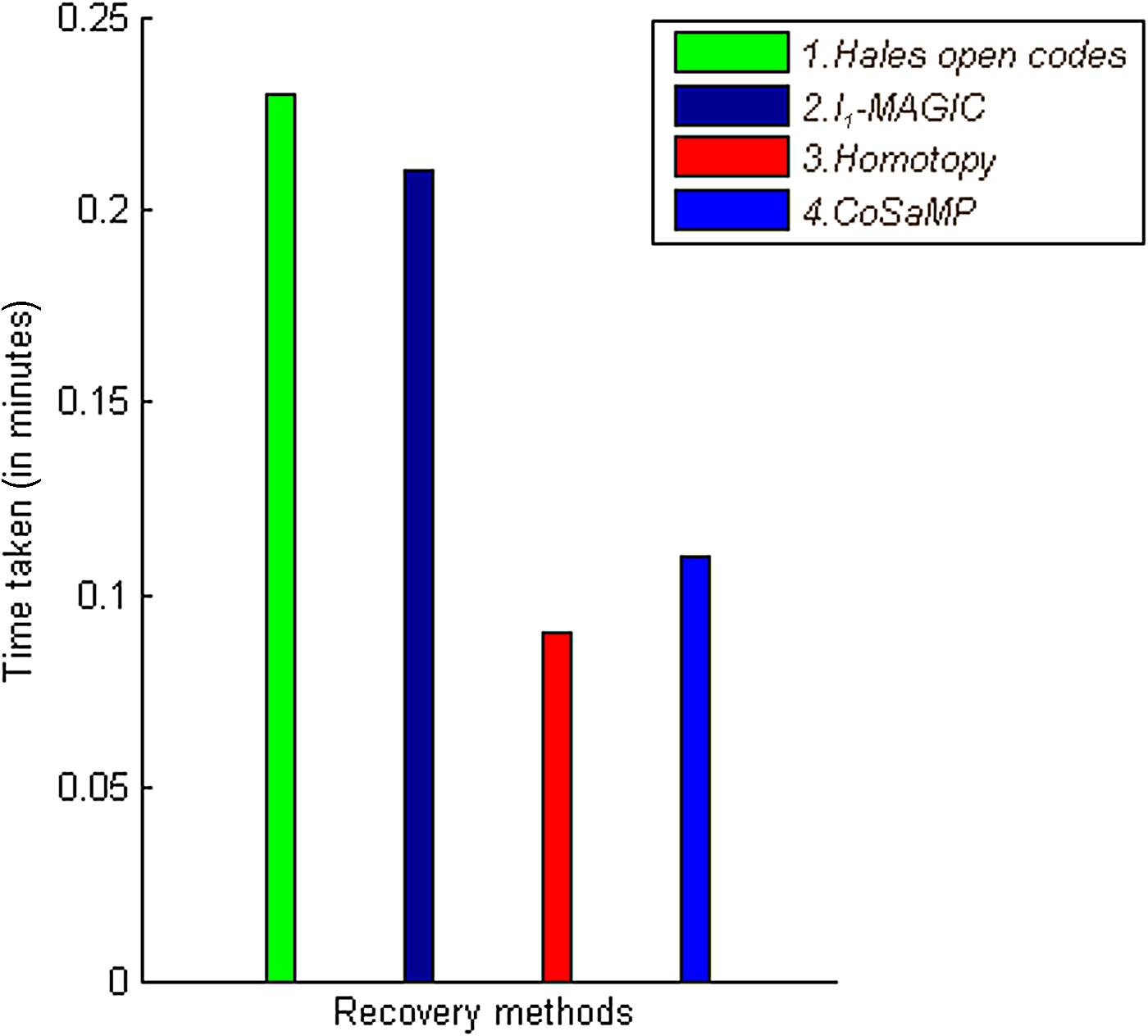
Comparison of recovery methods with respect to time. Computational time taken by recovery methods to recover a target concentration for EST datasets is compared.
Summary of the comparison of recovery methods in conjunction with different matrices of size 28 × 73 that can be used as binding patterns in selecting small interfering RNAs for EST dataset.
4.3. Metagenomics
The recovery methods are tested separately on microorganism genome datasets. Figures 7 and 8 show the performance of various recovery methods with respect to reconstruction error and time taken; as seen, l1-homotopy and CoSaMP perform better when compared to baseline GPSR and WGSQuikr recovery methods. It is also seen that CoSaMP reconstructs in much lesser time with closest approximations to higher concentration genera. The performance of l1-magic in terms of error decreases because the sensing matrix is not a positive definite matrix; hence, we have used a MATLAB script for solving this problem. Table 3 summarizes the results of metagenomics application, with concentration levels of top three genera.
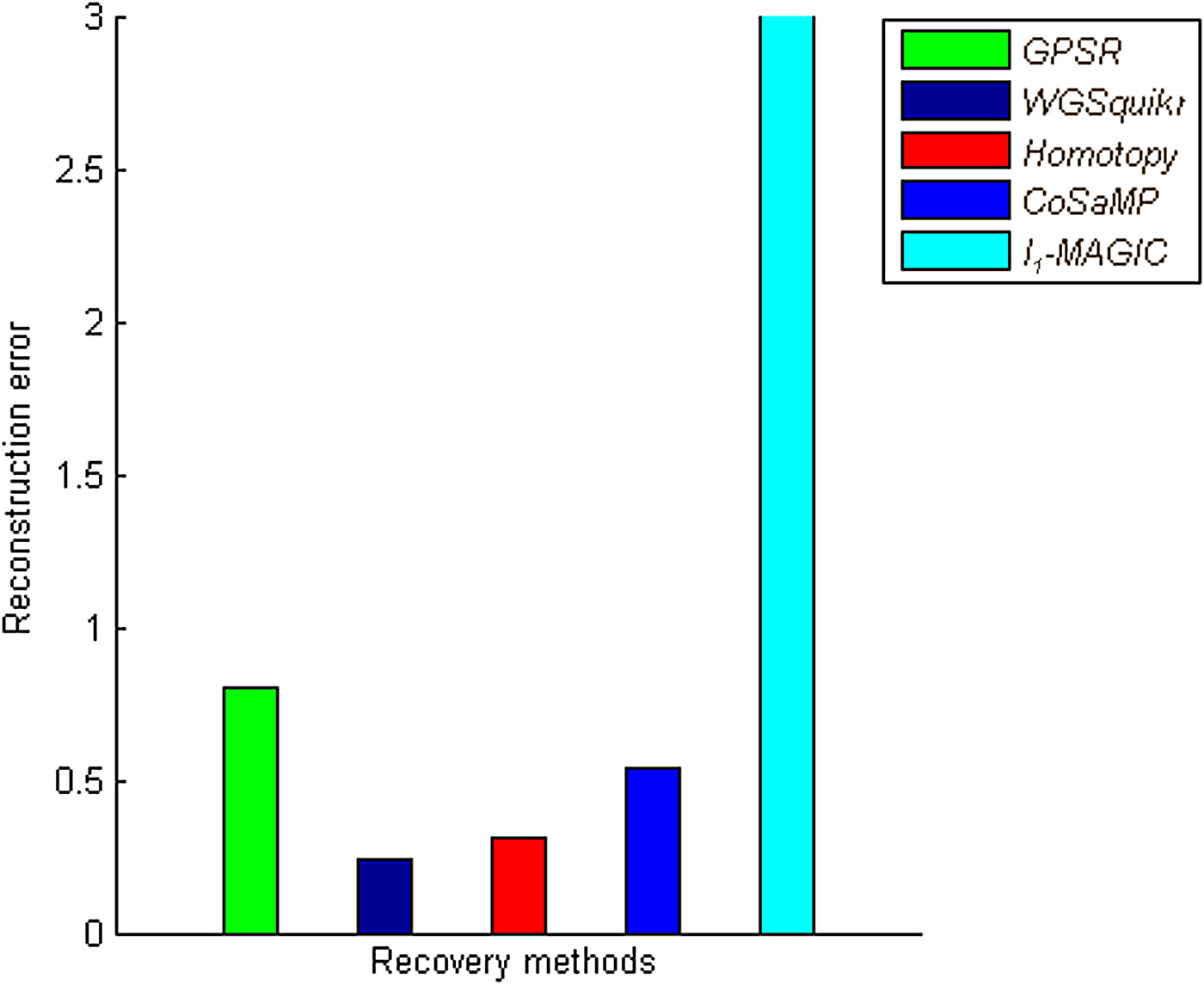
Comparison of recovery methods with respect to reconstruction error. Recovery methods are compared to baseline GPSR and WGSQuikr for large-scale bacterial genome dataset, and performance is analyzed in terms of l2-reconstruction error between the true abundance genera and reconstructed genera concentrations.
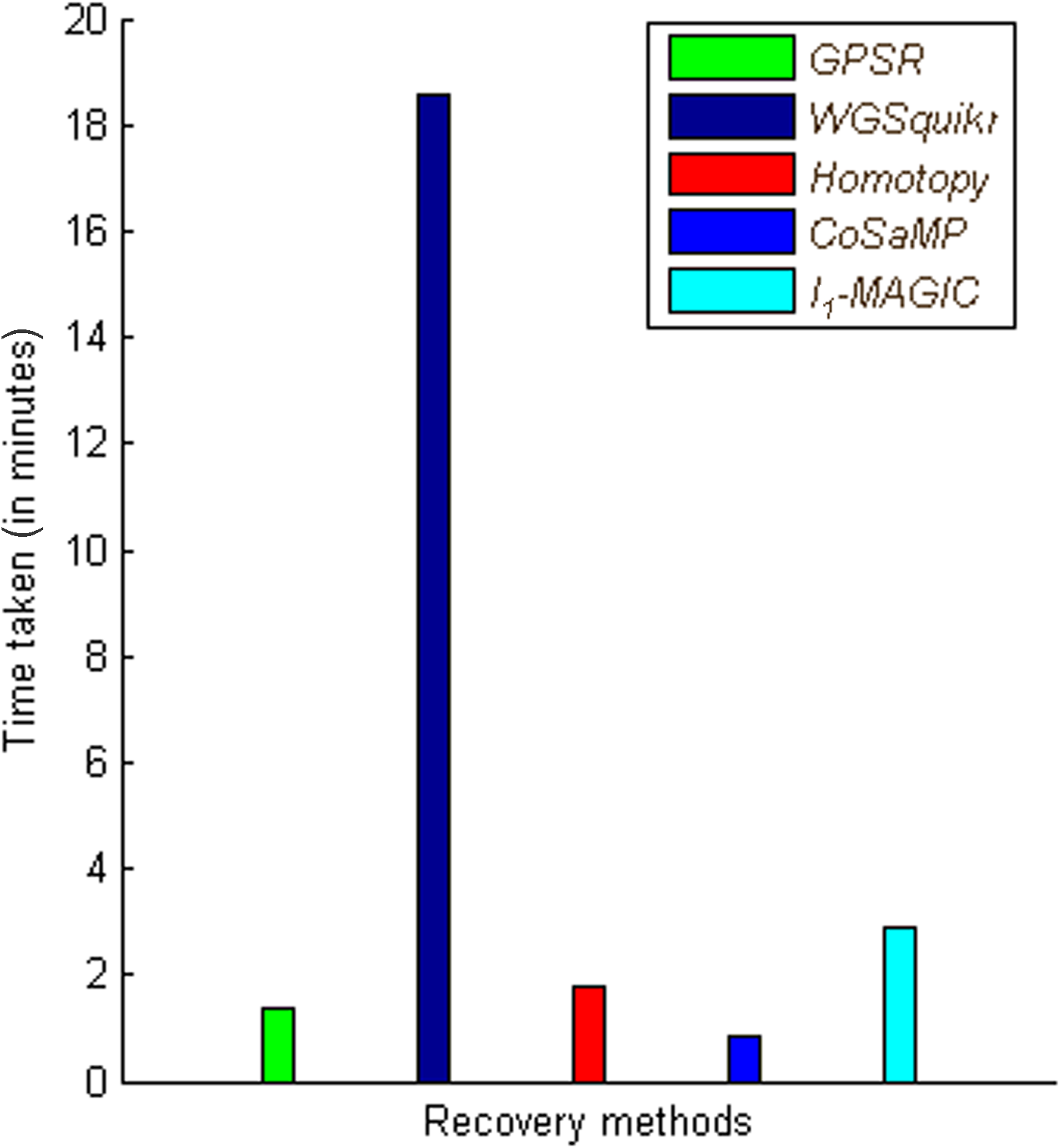
Comparison of recovery methods with respect to time. Time taken by the recovery methods to estimate the bacterial concentrations is compared for a large-scale genomic dataset.
Summary of comparison of recovery methods used in metagenomics application for whole-genome bacterial database.
5. Discussion
We have used three CS recovery methods, namely, l1-magic, l1-homotopy, and CoSaMP, for three genomic applications, namely, CSM, RNAi, and metagenomics. In each of these applications, previous work has shown how CS is relevant and that CS recovery methods significantly outperform the traditional methods, for both time and error. However, while it has been established that CS can successfully applied to genomics, a wider study of using different CS recovery methods was lacking.
We first applied CS recovery methods and matrices to real-world CSM datasets. We find that the l1-magic and l1-homotopy recovery methods show significant reduction in reconstruction error. However, if nonlinearity is considered in the measurement intensities, belief propagation continues to perform better. We then used the COG dataset for initial probe design and use large-scale EST datasets for selecting siRNA probes, although with an approximation of the binding pattern due to the limitation on the matrix size and available siRNA probes. We find that the l1-magic recovery method performs significantly better when compared to the baseline Hale's open codes method, with respect to both time and reconstruction error. We finally apply CS recovery methods in metagenomics in order to recover the target concentration of a bacterial genome dataset, and compare the results to the WGSQuikr method. It is seen that, in metagenomics, CoSaMP performs the fastest, while the l1-homotopy method shows significant reduction in reconstruction error.
In addition to improvements in time and error, there is one other advantage of using CS methods in genomic applications. The hardware requirements are modest, allowing for nearly constant execution time and low memory usage. This is particularly well-suited for analyzing very large datasets on a standard laptop computer. For this work, we have used a laptop having an Intel Core i5-2430M processor and 6 GB RAM, and running 64-bit Windows 7 Operating System.
A constraint of PG matrices is that all sizes are not available. In fact, due to nonavailability of 3 × 7 and 48 × 146 PG matrices, we had to use 4 × 7 and 28 × 73 matrices for the COG sequences and the real-world EST dataset, respectively. It has been reported that it is possible to vary the size of the PG matrix by puncturing (Tian and Jones, 2005). We propose to try and use this procedure so as to arrive at optimum PG matrices. Certain applications require that the sensing matrix be positive definite. For the metagenomics example, we intend train matrix to be positive definite so as to get better estimation of concentration levels of targets. Finally, we also intend to explore the usage of Euclidian geometry matrices of different sizes (Attili, 2006).
6. Conclusions
The principle of using different CS recovery methods in various genomic applications has previously been established, but a comprehensive study of using a wide range of CS methods had not been done. In this work, we successfully used different CS recovery methods for genomic applications. As a novelty, we considered PG-based matrices and compared these against the performance of baseline matrices. Significant reduction in reconstruction error as well as execution time was observed. The l1-homotopy reconstruction method stands out from the other recovery methods as it performs well in all three applications. A key differentiator of this work is the usage of real-world datasets, thus ensuring scalability.
Footnotes
Acknowledgments
We are grateful to various groups who have made available codes and implementations for various recovery methods, as part of their publications. This includes l1-magic (Candès and Romberg, 2005), belief propagation (Sarvotham et al., ![]() ), and so on.
), and so on.
All authors conceptualized the work and gave the inputs for the various modules of the tool. D.P. involved in the development of the entire components and in drafting the article. A.R. helped in the biological aspects and helped in drafting and revising the article. C.H.R. reviewed the results. R.S. reviewed the work and gave biological insights. All authors read and approved the final article. We thank Dr. Thomas Joseph for his valuable comments on the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.