
Abstract
Abstract
As widely discussed in literature, spatial patterns of amino acids, so-called structural motifs, play an important role in protein function. The functionally responsible part of proteins often lies in an evolutionarily highly conserved spatial arrangement of only a few amino acids, which are held in place tightly by the rest of the structure. Those recurring amino acid arrangements can be seen as patterns in the three-dimensional space and are known as structural motifs. In general, these motifs can mediate various functional interactions, such as DNA/RNA targeting and binding, ligand interactions, substrate catalysis, and stabilization of the protein structure. Hence, characterizing and identifying such conserved structural motifs can contribute to the understanding of structure–function relationships. Therefore, and because of the rapidly increasing number of solved protein structures, it is highly desirable to identify, understand, and moreover to search for structurally scattered amino acid motifs. This work aims at the development and the implementation of a novel and robust matching algorithm to detect structural motifs in large sets of target structures. The proposed methods were combined and implemented to a feature-rich and easy-to-use command line software tool written in Java.
1. Introduction
1.1. Biological relevance
T
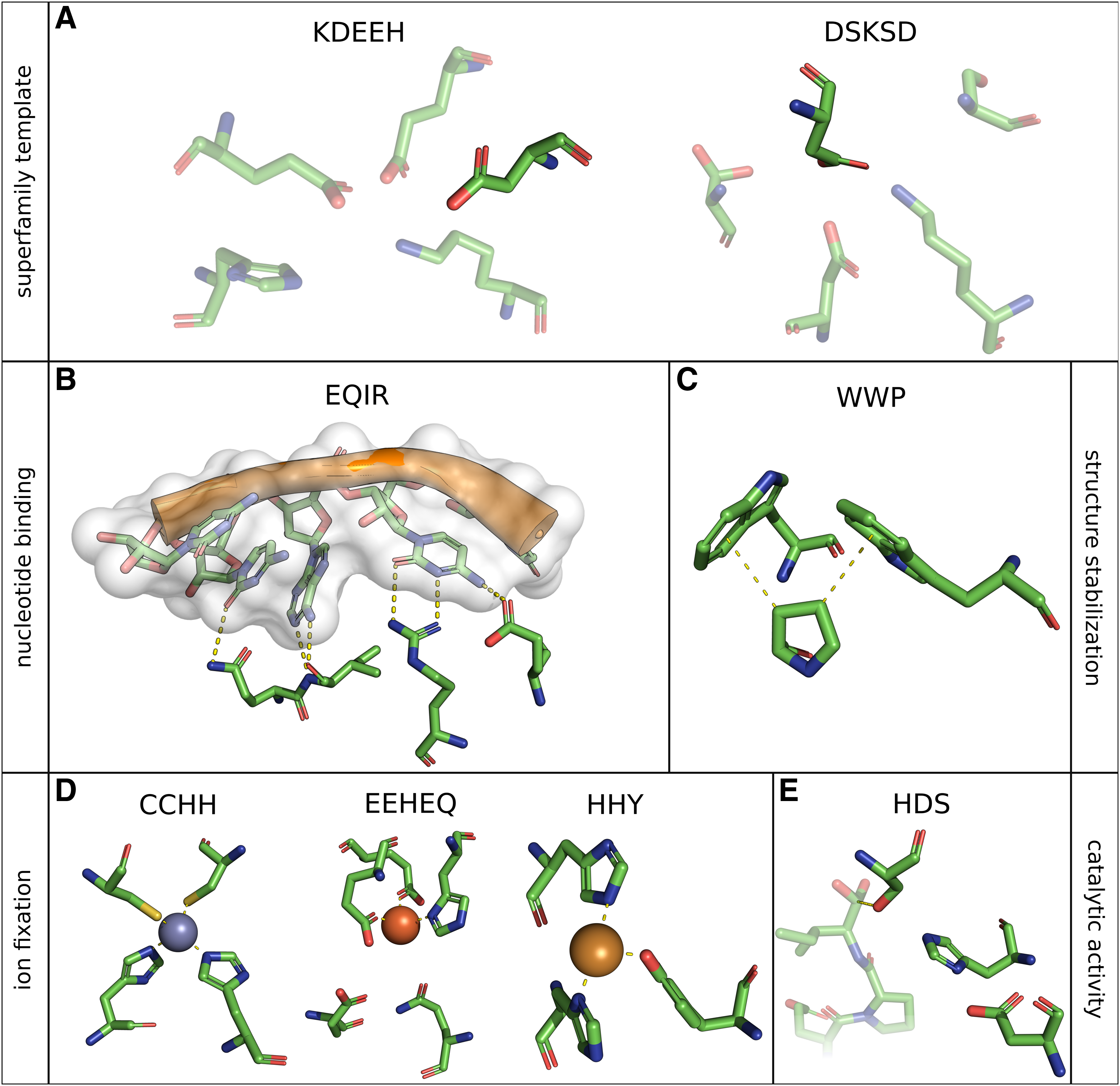
Selection of biologically relevant motifs. (
Shown for each motif are nomenclature, type, short description, source structure, chain(s) of occurrence, incorporated residues in the format [chain ID]-[amino acid type][residue number]→[exchange] and reference.
For instance, the ability to degrade polypeptides is an essential biological process. This chemical reaction is usually catalyzed by certain substrate-specific enzymes, the proteases. The first identified protease catalytic site was revealed in 1967 by studying α-chymotrypsin with X-ray diffraction (Matthews et al., 1967). Subsequent structure alignment of proteases uncovered remarkable similarity of active sites (Fischer et al., 1994). It was found that a spatial arrangement of three amino acids, a so-called triad, consisting of histidine, aspartate, and in most cases serine (Fig. 1, HDS), is responsible for peptide bond cleavage in proteases. For the purpose of conservation and structure stabilization the catalytic triad is part of an extensive hydrogen bonding network (Hedstrom, 2002). It was reported that, in general, serine attacks the carbonyl group of the peptide bond whereby histidine acts as general base in the first step. The protonated histidine is then stabilized by formation of hydrogen bonds with aspartate. Finally, by involving water, the amino group is broken (Hedstrom, 2002). Serine proteases are involved in many in vivo reactions like complex cascading inflammation processes, vascular homeostasis, thrombosis (Sharony et al., 2010) or chronic pancreatitis (Witt et al., 2000). Furthermore, decreased levels of the serine protease neurosin were observed in brain tissue of patients suffering from Alzheimer's or Parkinson's disease, although direct linkage is still unknown (Ogawa et al., 2000). Additionally, recent studies show evidence that selected inhibition of human immunodeficiency virus associated serine proteases can stop the spreading of the infection by preventing cleavage of viral polyproteins in functional subunits and hence thwart virus maturation (Titanji et al., 2013). Consequently proteases are highly important drug targets (Sanderson, 1999), thorough understanding of their molecular structure is mandatory, and bioinformatic investigation of catalytic mechanisms broken down to structural motifs is worthwhile.
Additional recently discussed topics include Me/π and XH/π interactions induced by aromatic amino acids. Widely present in proteins, such interaction motifs are evidently essential for protein function and folding (Plevin et al., 2010). In this context Koutsotoli and Tzakos described the disruption of the protein–protein interaction network in human host cells by infection of enterohemorrhagic Escherichia coli (EHEC). Here a π-π stacking interaction of two tryptophan residues sandwiching a proline is exploited during EHEC infection process. This so-called C-H-π interaction motif (Fig. 1, WWP) was found to be present in more than 600 cases in the PDB (Koutsotoli and Tzakos, 2012).
Furthermore, the active binding site of human H-chain ferritin (PDB:1FHA) that is essential for bioavailable iron storage (Ebrahimi et al., 2012) can be described as a structural motif consisting of five residues mediating ion binding (Fig. 1, EEHEQ).
These are only a few examples of functionally tailored, highly specific and conserved structural motifs that occur among different protein families and thus led to proteins that share common functional characteristics by evolutionary means. Moreover, the representation through structural motifs can be adduced to describe classes of proteins. Function can be preserved during protein evolution despite overall sequence identity or fold shape diverges and even if common function is not obvious, partial reactions can be shared. The motif KDEEH (Fig. 1, KDEEH), based on a mandelate racemase structure (PDB:2MNR), was derived by Meng et al. in 2004 and comprises several structures of the enolase superfamily (ES). Active site exchanges happened during evolution of the ES, nevertheless a partial chemical reaction as common function was persistent: the support of proton abstraction from carbon adjacent to carboxylic acid and the subsequent formation of an enolate anion intermediate (Babbitt et al., 1996). The lysine residue at position 164 can be substituted by histidine, glutamate 247 by aspartate or asparagine, and histidine 297 by lysine. Using this template definition it was possible to represent the ES and their subgroups appropriately (Meng et al., 2004). Another example of a superfamily representing motif is DSKSD derived from PDB:1QQ5 (Fig. 1, DSKSD). Like the ES, the haloacid dehalogenase superfamily (HADS) is thought to have evolved with a partial reaction constrained: the catalysis of hydrolytic nucleophilic substitution resulting in a covalent bond between a fully conserved aspartate and an atom of the substrate (usually phosphate) (Meng et al., 2004). Likewise to KDEEH, residue definitions are not stringent and substitutions are permitted (Table 1, DSKSD).
1.2. Motivation
On the one hand computational examination of substructure similarities is highly desirable, but on the other hand existing methods are limited or deprecated. Table 2 provides a brief overview of existing approaches, their underlying methodology, availability, and purpose of application. In literature amino acid representation of protein structures in the context of substructure matching was mainly focused on single or a few atoms like C α -only, C α and C β , or pseudo-atom-based sidechain representations (Debret et al., 2009; Moll et al., 2010). While the latter represents—even if only marginal—the different types of amino acids geometrically, C α -only representation does not allow the geometric mapping of amino acid types. A pseudo-atom sidechain representation as compromise may provide a good trade-off between fuzziness and sensitivity of the search method and is less sensitive to conformational changes (Meng et al., 2004). But the fact that such representations are insufficient and inaccurate in some cases was proven, for example, for the far-reaching ES (Meng et al., 2004). This carries great weight if proteins are of limited sequence identity and hence structural variant, which is known for ES (Meng et al., 2004; He et al., 2013) and even more evident for HADS (Meng et al., 2004; Aravind et al., 1998; Koonin and Tatusov, 1994; Baker et al., 1998).
An overview of existing approaches for structural motif searching showing their underlying method, availability as web service (WS) or standalone tool (ST), application, and reference.
Exploring the effect of considering specific atoms for matching, which might be important for or directly involved in active site functionality, is highly desirable and limitations of previous methods were already recognized (Meng et al., 2004). A holistic representation that allows for incorporating all atoms or arbitrary subsets of atoms can form a case-dependent and adequate mapping of functional important or structure-forming elements. Hence, detailed definition of atom representation is mandatory to retain specificity in certain cases. Nonetheless previous software lacks such arbitrary atom definitions (Debret et al., 2009; Moll et al., 2010; Nadzirin et al., 2012) and consequently this hurdle has to be overcome.
Furthermore, structural motifs can occur in the same protein chain (intramolecular) or scattered among different protein chains (intermolecular) per se (Koutsotoli and Tzakos, 2012; Ekici et al., 2008; Tsukada and Blow, 1985). Although intramolecular occurrences are more common, intermolecular motifs are often observed in protein–protein interfaces for structure stabilization (Koutsotoli and Tzakos, 2012), and ligand and substrate binding (Ekici et al., 2008), as well as contact sites in general. Therefore intermolecular findings are highly important, albeit most available matching methods are limited to single chains (Debret et al., 2009; Nilmeier et al., 2013; Konc and Janezic, 2010).
The problem to find a set of geometrically and compositionally defined amino acids (query motif) in a set of target structures is rather complex. The geometric component of the problem can be defined as pattern-matching in the three-dimensional space, while the compositional component introduces further restrictions regarding the amino acid types matched (He et al., 2013). These can be for instance the requirement of exact amino acid matching or the toleration of chemical similar or arbitrary residue substitutions. The compositional constraint can even be further tightened such that amino acid substitutions are restricted to specific positions of the motif and not, for instance, the exchange of tryptophan to glycine in general. This gains importance if structural motifs are considered that contain more than one amino acid of the same type (WWP, Fig. 1). Hereby it is essential to distinguish between substitutions at different residue positions. Exchanges of the tryptophan residue oriented toward the nitrogen and the C
δ
atom of proline are not necessarily identical to those of the tryptophan facing proline C
β
and C
γ
atoms. Here so-called position-specific exchanges (PSEs) are crucial and of high interest to restrain structure-disrupting substitutions during matching, especially if motifs are occurring inter molecularly. However, nearly in all current matching methods the implementation of PSEs is lacking (Nadzirin et al., 2012; He et al., 2013; Debret et al., 2009; Kleywegt, 1999; Konc and Janezic, 2012). Consequently the outlined drawbacks were tackled during development and particular attention was paid to:
• arbitrary and user-definable atom representation of motifs, • detection of intra- and intermolecular matches, • and the definition of PSEs.
The lack of an easy-to-use implementation of a structural motif search algorithm underscores the necessity to develop versatile tools suitable for contemporary high-throughput analyses. Nowadays the number of protein-related data increases rapidly and structures are released prior to biochemical and functional annotation due to structural genomics effort and automated structure determination pipelines (Duarte et al., 2012). Hence, screening against libraries of structural motifs, for instance, derived from the Catalytic Site Atlas (CSA) (Torrance et al., 2005), may reveal hidden functional aspects. Furthermore, the identification of a partial chemical reaction for protein superfamilies may be possible and can aid deducing a superfamily representative template (Meng et al., 2004). As a consequence, spatial patterns of amino acids that are directly representing structure–function relationships can be utilized to search databases of protein structures with high sensitivity and allow for uncovering substructure similarity of divergently evolved proteins that are likely to share function (Meng et al., 2004; He et al., 2013). Therefore, the utilization of search methods for structural patterns can aid researchers to identify possible targets for drug repositioning or to discover off-target binding of specific drugs (Kirshner et al., 2013; Xie et al., 2009). Where similar substructure fold is observed it is possible that there are drug-binding capabilities as well, causing unwanted side effects. Additionally, the detection of allosteric protein-binding sites is a reasonable application of structural motif search algorithms. Newly determined or even theoretical (e.g., homology modeling based) structures can be screened against a library of binding sites or other relevant motifs to find and further investigate potential new drug targets (Kirshner et al., 2013).
1.3. Nomenclature
A unified nomenclature is used to increase comprehensibility of the following elucidations and to define an unmistakable description for structural motifs. At the motif level, one-letter codes of motif-incorporated amino acids are concatenated following their ascending sequential order. Unambiguous amino acids are labeled consistently with the chain of occurrence, one-letter notation of amino acid type and residue number according to the corresponding Protein Data Bank (PDB) entry.
For example, consider the widely discussed structural motif present in zinc finger domains, which is constituted of a pair of cysteine residues and a pair of histidine residues each (Miller et al., 1985). This motif, derived from PDB:1G2F, consists of the residues F-C207, F-C212, F-H225, and F-H229 and is therefore denoted as CCHH.
If PSEs are allowed they are indicated by an arrow following the sequence number of the residue that should be declared as a variable. The ES representing motif constituted of lysine, asparagine, two glutamate residues, and histidine (Meng et al., 2004) was derived from PDB:2MNR and is denoted as KDEEH according to the sequential order of the incorporated amino acids: A-K164 → H, A-D195, A-E221, A-E247 → DN, A-E297 → K.
1.4. Substructure similarity
For the assessment of geometric resemblance of structural motifs and a set of substructural match candidates of a target structure, the least-root-mean-square deviation (LRMSD) is used. The LRMSD (Eq. 1) is defined as the minimal root-mean-square deviation (RMSD) (Eq. 2) over all possible ideal superimpositions computed by singular value decomposition (Golub and Reinsch, 1970) of all permutations of two sets of atoms A and B (Fofanov et al., 2008). Hereby only atoms of the same type are considered for pairwise alignment to reduce computational effort.
However, inferring geometric similarity alone does not give a clue on significance of hits in a statistical and functional meaning (Fofanov et al., 2008). The LRMSD can vary greatly, depending on the number of atoms considered for calculation and the type of amino acids compared (Stark et al., 2003). Therefore the LRMSD is not suitable to infer functional similarity and, further, it is not trivial to define a universal significance threshold (Fofanov et al., 2008). For matches incorporating allowed PSEs, and if atoms beyond C α are considered, there is a limitation for geometric similarity computation: comparison of different residues is futile. Hence a simplification was introduced that considers only C α atoms for alignment of different amino acid types, despite definition of which atoms should be used for alignment. However, this exception only applies when PSEs are defined and hence direct comparison of nonidentical residues is mandatory.
To get rid of the incomparability of LRMSD values several methods were presented to apply a statistical model to estimate match significance (Moll et al., 2010; Stark et al., 2003; Fofanov et al., 2008; Xie et al., 2009). In this work a point-weight corrected model originally developed by Fovanov et al. was used to estimate significance and to diminish the influence of runtime-beneficial geometric constraints (e.g., LRMSD cutoff) by maximum likelihood approximation (Fofanov et al., 2008).
2. Methods
2.1. Algorithm
The following elucidations are supported by Figure 2, which illustrates the spatial abstractions used in the approach. Furthermore, Algorithm 1 shows the corresponding pseudo code listing of the search algorithm.
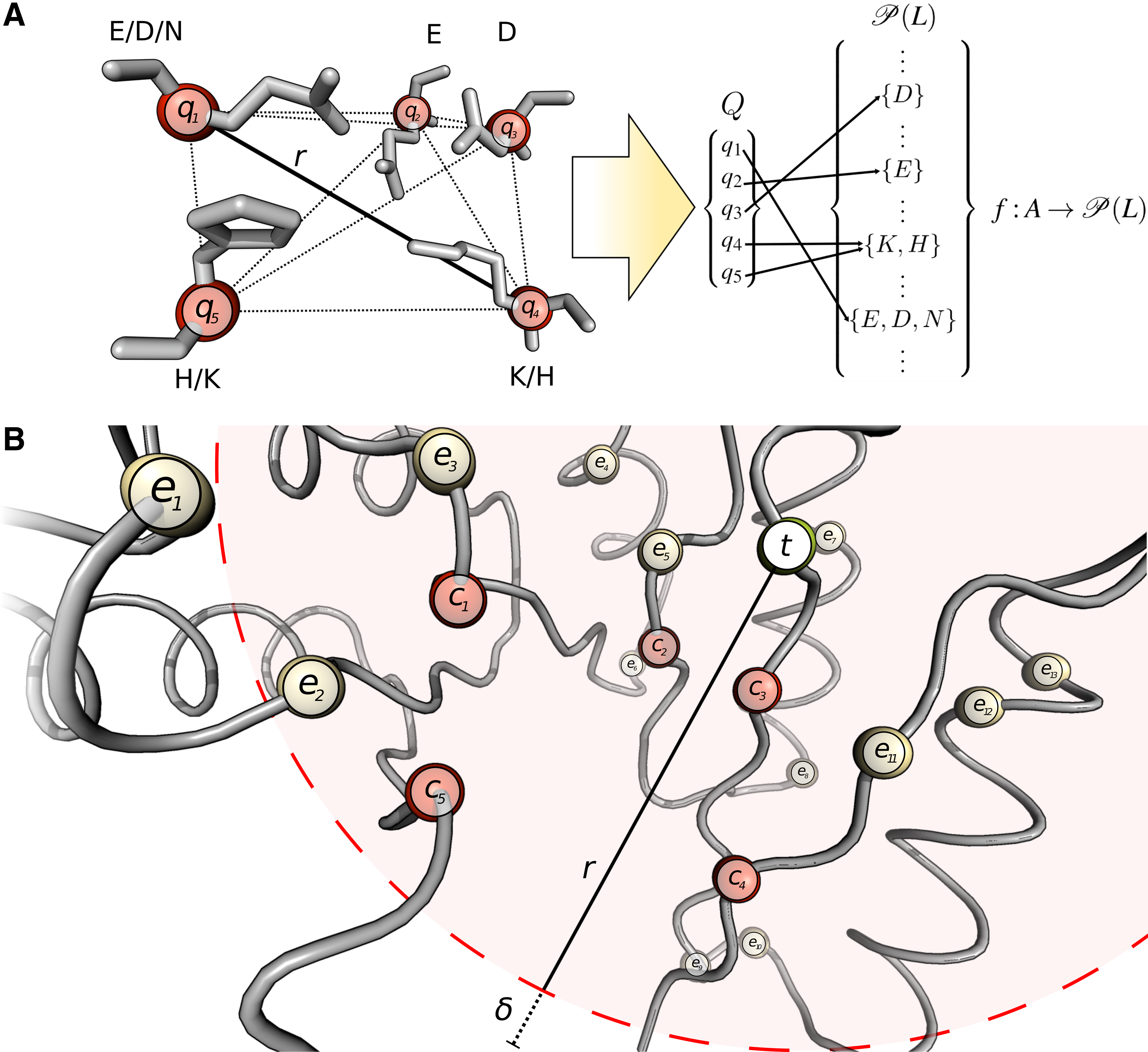
Exemplary illustration of the Fit3D algorithm for motif KDEEH. (
The set
The search itself is an iterative process over all points in the target set T (Algorithm 1, line 7). If g (t) is a subset of at least one set of allowed amino acid labels for each
If pairwise distance filtering of Et is enabled, only pairs of
For each combination C where |C| = k and compatibility of amino acid labels is given according to
2.2. Time complexity
The motif spatial extent r can be found in time

However, it has to be admitted that the complexity of the Fit3D motif matching algorithm strongly depends on the density and size of the local environment. For target amino acids t buried in the protein, l tends to be bigger than for target amino acids at the protein surface and vice versa. In the worst case l could be as large as n, which happens practically never because the definition of small and locally occurring structural motif implies that l ≪ n.
2.3. Algorithm evaluation
For validation of the presented algorithm a two-step gold standard was defined to determine coverage of a foreground and background benchmark data set. The validation of substructure matching algorithms based on ES structures and CSA derived motifs was successfully applied several times in literature (Moll et al., 2010; He et al., 2013; Fofanov et al., 2008). Based on the assumption that statistically significant matches can be considered to be true positives, validation was conducted on the basis of p-value estimation of hits with a well-established cutoff of 0.001 (Moll et al., 2010; Moll and Kavraki, 2008). The limiting geometric threshold ε that defines the maximal allowed LRMSD was set to 4.0 Å to cover a good proportion of the right tail of the LRMSD distribution. In all, 31.133 structures of the nonredundant PDB (nrPDB) chain set as of March 14, 2014, with a BLAST p-value of 10−80 (Altschul et al., 1990) were considered as background dataset. Furthermore, the set of 41 structures by Meng et al. (Meng et al., 2004) was used as foreground for validation based on the ES. Additionally, ES-based evaluation was conducted in dependence of different representations for motif KDEEH to show crucial influence on sensitivity and specificity: all non-hydrogen atoms, backbone atoms, sidechain atoms, and C
α
atoms. All CSA-derived motifs were considered to be representatives and used for evaluation if each of the following conditions was fulfilled (adapted from Moll et al., 2010):
• the motif consists of 3–5 residues, • the parent structure is fully classified by the Enzyme Commission (EC) number, • the corresponding EC class has more than 50 structures, • and the motif has a maximal spatial extent of 20 Å.
Employing these filter criteria for selection yielded a total number of 157 CSA-derived motifs where p-values could be computed, spanning 51 different EC classes and all six top-level classifications.
2.4. Implementation
The open source and platform-independent Java implementation of the algorithm was aimed to be simple in usage and flexible in application. Hence, the decision to deploy the software as command line tool considerably reflects these aspects. Extensive usage of the BioJava (Prlic et al., 2012) framework allowed to circumvent de novo implementation of PDB file parsers and structure alignment methods. The Fit3D algorithm implementation was realized by utilization of the BioJava data structure. Hence, special attention could be paid to runtime optimization.
3. Results and Discussion
3.1. Algorithm evaluation
The results of algorithm validation with motif KDEEH are shown in Figures 3 and 4. All-atom as well as sidechain representation resulted in perfect coverage of the foreground dataset (sensitivity of 100%), which dropped significantly if only backbone atoms were used as motif representation. Less than half of the foreground structures were identified if only C α atoms were used for matching. A similar behavior was observed for specificity, which is even slightly better for all-atom representation (99.68%) than for sidechain-based motif abstraction (98.11%). For backbone-only representation of KDEEH a significant loss of sensitivity was observed (85.37%) that was even higher if only C α atoms were considered. In general, all-atom motif mapping performed best, followed closely by sidechain representation. In contrast, C α representation performed worst for all measurements with extraordinary low sensitivity and specificity.
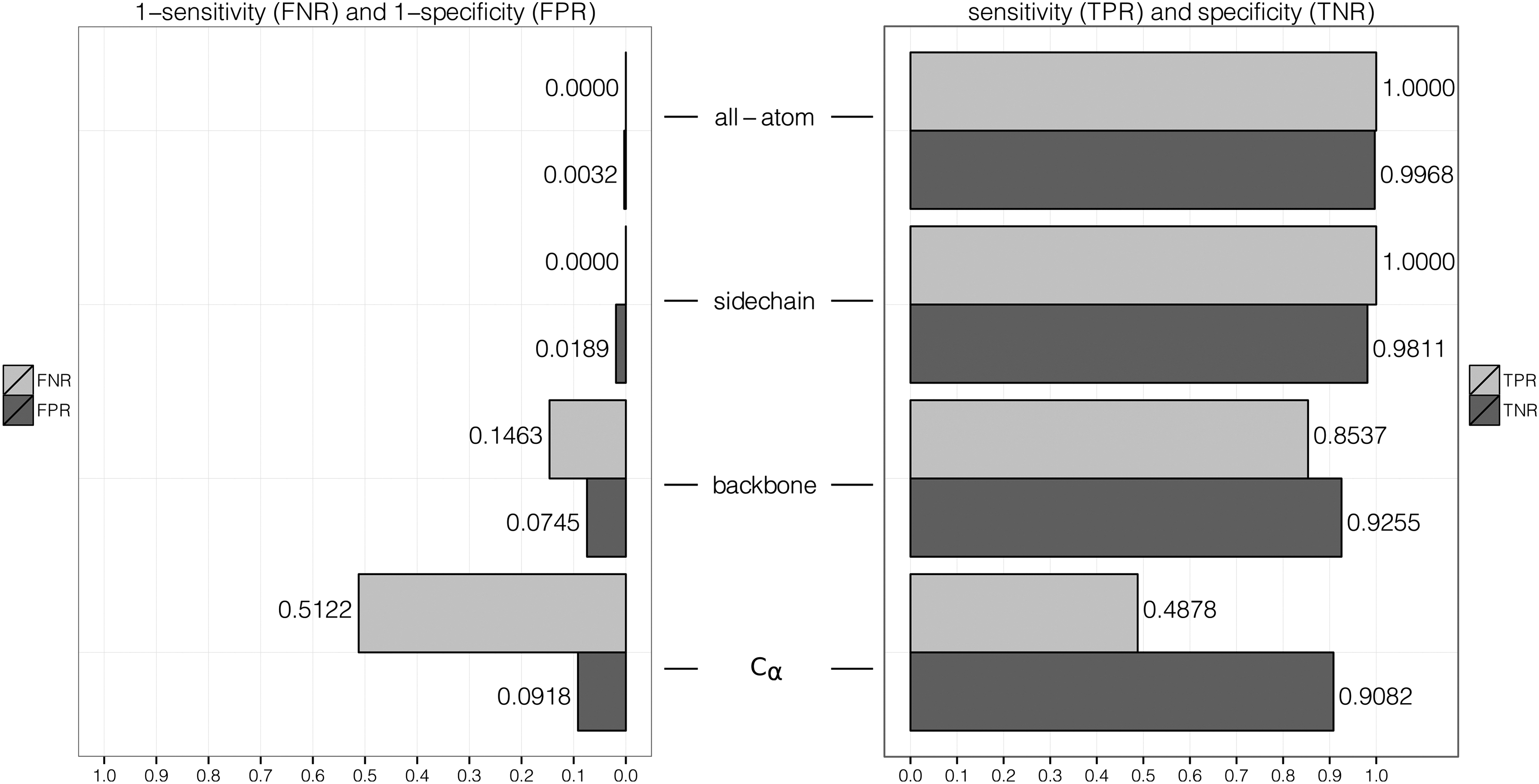
Enolase superfamily algorithm validation results. The sensitivity or true positive rate (TPR) and specificity or true negative rate (TPR) as well as their inverse measurements false negative rate (FNR) and false positive rate (FPR) are shown in dependence of atom representation for the enolase superfamily motif.
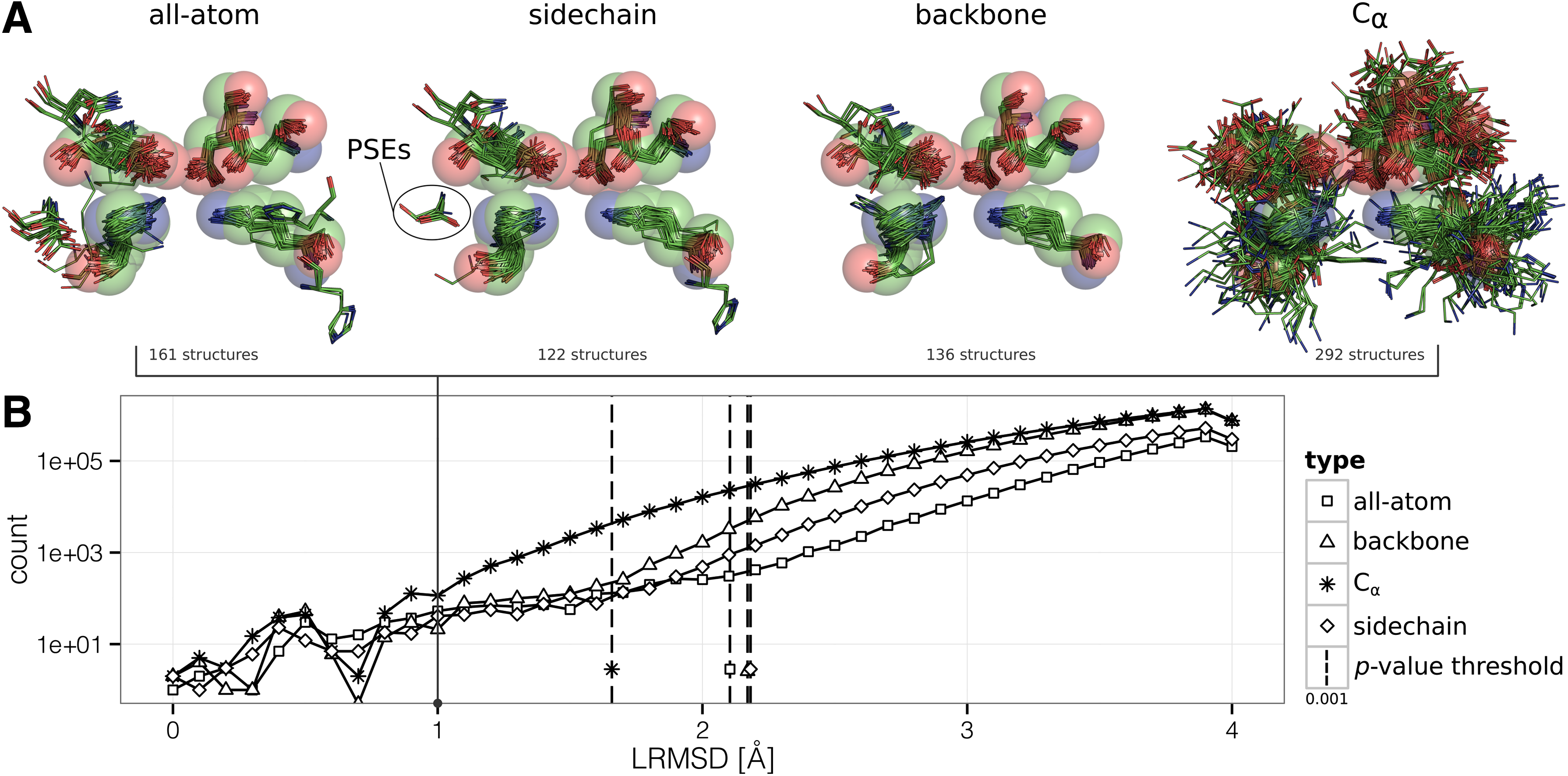
Influence of motif representing atoms. (
By comparison of the false positives identified according to the dataset defined by Meng et al. in 2004, with the up-to-date set of ES structures derived from the Structure Function Linkage Database (SFLD) (Akiva et al., 2014), it is even possible to assess some false positives as true positives. The discrepancy in size of the two datasets is remarkably 41 structures for Meng et al. (2004) compared to 351 structures in SFLD as of June 10, 2014. For example, the structures PDB:4GFI or PDB:3RIT, which were matched as false positives, have now been identified to belong to the ES like reported by the SFLD. In contrast to 99 false positives regarding the Meng et al. (2004) dataset, only 40 remain if the SFLD dataset is considered as foreground, increasing the method specificity slightly from 99.68% to 99.87%. Additionally, the structures PDB:2QGY, PDB:2PPG, PDB:2OQH, PDB:3CYJ, PDB:1WUE, PDB:2OZ8, and PDB:2POZ have indeed only putatively defined functions, but may be related to the ES (Moll et al., 2010). In fact, these results are a first indicator for the ability of the proposed search algorithm to identify remote homologous structures.
A frequency plot showing LRMSD distribution in dependence of motif representation is given in Figure 4B. The corresponding alignments of sets of KDEEH matches with similarities to the query motif ≤1.0 Å illustrate different motif representations in Figure 4A. As expected, the LRMSD distribution for all representation types has a smaller peak in the left region (see Fig. 4B) and diverges at the right tail in accordance to observations of previous studies (Fofanov et al., 2008; Moll et al., 2010; Stark et al., 2003). Furthermore, the corresponding LRMSD at the p-value threshold of 0.001 is shifted toward a lower value if reduced and insufficient atom sets are considered: the nonspecificity is directly reflected in LRMSD distribution and thus also in the statistical significance. This is also perceptible by a closer look at Figure 4A, where the query motif KDEEH is shown as sphere model with a radius of 1.0 Å for each sphere. The sphere model intends to illustratively define the area of atom deviation from match to query, which is enormously for C α -only representation, despite all matches are still below significance threshold. In contrast, match deviations are significantly lower for other representation types with no obvious differences at first glance. Note that outlying amino acids occur due to allowed residue substitutions (PSEs) of KDEEH and contributed to LRMSD calculation only with their C α atoms.
It was clearly shown that atom representation of structural motifs plays a key role when it comes to matching in target structures. It was expected that multi-atom motif representation performs best, as already suggested for the ES (Meng et al., 2004). The unavailability of arbitrary atom selection is a known deficiency of existing approaches, and with our program it could be addressed appropriately. For the ES, which is known to have highly variable C α atom positions, all-atom representation performed best, although the performance for sidechain motif abstraction was close. Both variants reached a perfect sensitivity and a nearly ideal specificity, albeit sidechain representation led to a slight loss of specificity compared to all-atom representation. Further on, backbone-only matching lags behind with a still acceptable but not desirable sensitivity, while C α -only abstraction showed its strong inaccuracy.
In conclusion it was confirmed that multi-atom representation is the method of choice, highly important, and a worthwhile key feature of the Fit3D software. Albeit this statement cannot be generalized for every motif and has to be reflected for each specific search task. For instance, if variable regions of the motif are known or positional variance is even favored, it may be reasonable to constrain atom representation.
The results of the algorithm validation using CSA-derived motifs are shown in Figure 5. The CSA motifs were represented by all non-hydrogen atoms, but the results were rather diverging. While the sensitivity ranged from 0–95.60%, specificity was continuously high, which means that no excessive matches in the background dataset occurred. If we consider for example the best performing motifs derived from PDB:1STC (cAMP-dependent protein kinase, EC 2.7.11.11), PDB:1BSJ (peptide deformylase, EC 3.5.1.88), PDB:1BBS (renin, EC 3.2.23.15) and PDB:1AMY (α-amylase, EC 3.2.1.1), it is conspicuous that three of them span the same EC top-level class (hydrolases, EC 3). More detailed investigation reveals that the CSA entries derived from PDB:1BBS and PDB:1AMY are both catalytic triads consisting of two aspartate residues and one glutamate (PDB:1AMY) or serine (PDB:1BBS). According to SFLD α-amylase (PDB:1AMY) is known to catalyze hydrolysis of internal α-glucosidic linkages in starch and other related oligo- and polysaccharides (Furnham et al., 2014). Renin (PDB:1BBS) instead catalyzes, according to the ENZYME database (Bairoch, 2000), the cleavage of Leu-Xaa (Xaa can be any amino acid) bonds in angiotensinogen. Hence, the catalytic triads seem to be very descriptive for their EC class and therefore highly conserved. Other CSA-based motifs like the aspartate, lysine and tyrosine triad from fructose-bisphosphate aldolase (PDB:1OK4, EC 4.1.2.13), were not even able to detect one hit in the respective foreground dataset. We suppose that observed divergence in the performance of CSA-derived motifs is induced by unknown PSEs. This issue could be addressed if multiple sequence alignments of proteins in the motif-associated EC class are performed as already suggested in literature (Moll et al., 2010; He et al., 2013). Otherwise hand CSA-derived substructures are not per se structural motifs in a common sense. The CSA developers did not intend to define motifs descriptive for EC classes, nor the CSA was designed to cover all EC classes (Torrance et al., 2005). Instead, the goal to allow functional annotations of disparate proteins led to CSA development (Torrance et al., 2005). Torrence et al. considered the foreground for each motif to be the “CSA family” of PSI-BLAST (Altschul et al., 1997) identified relatives, rather than the full fourth-level EC classification. Furthermore, a single EC class is likely to have more than one representative CSA entry, which are highly different concerning their descriptive abilities. Obviously this is the case for the entries PDB:1BP2 and PDB:1CJY, both catalytic sites of phopholipases (EC 3.1.1.4). While entry PDB:1BP2 is a catalytic triad of histidine, glycine and aspartate, the active site of PDB:1CJY consists of four amino acids: serine, two glycine residues and an aspartate. Although both are catalyzing the same reaction, the cleavage of ester bonds, they are very variant concerning the ability to describe their EC class and consequently the applied validation approach fails. The triad derived from PDB:1BP2 reached a sensitivity of 71.48% and specificity of 99.37%, while CSA entry PDB:1CJY only covered 0.39% of the foreground and was to 99.55% specific. These hurdles concerning CSA-derived motifs were already part of research and some suggestions to overcome them were presented (Bryant et al., 2010); Chen et al., 2007). One idea is the combination of multiple motifs describing one EC class to increase performance. Bryant and colleagues introduced a method in which multiple and potentially overlapping motifs can be combined to a single function prediction test (Bryant et al., 2010). Another approach encompasses the further optimization of motifs by geometric sieving (Chen et al., 2007), something of which a preliminary variant was already implemented in LabelHash (Moll et al., 2011). The application of these methods could help to increase the representative level of CSA-derived structural motifs and should be considered in latter work. All in all, the validation results for the ES-descriptive motif KDEEH and the CSA library showed that clearly and meticulously defined motifs that were derived from literature-supported superfamily structures are more descriptive for EC classes and hence these motifs can be considered to be of high quality. This corresponds to observations that were proposed by previous studies (Moll et al., 2010; He et al., 2013).
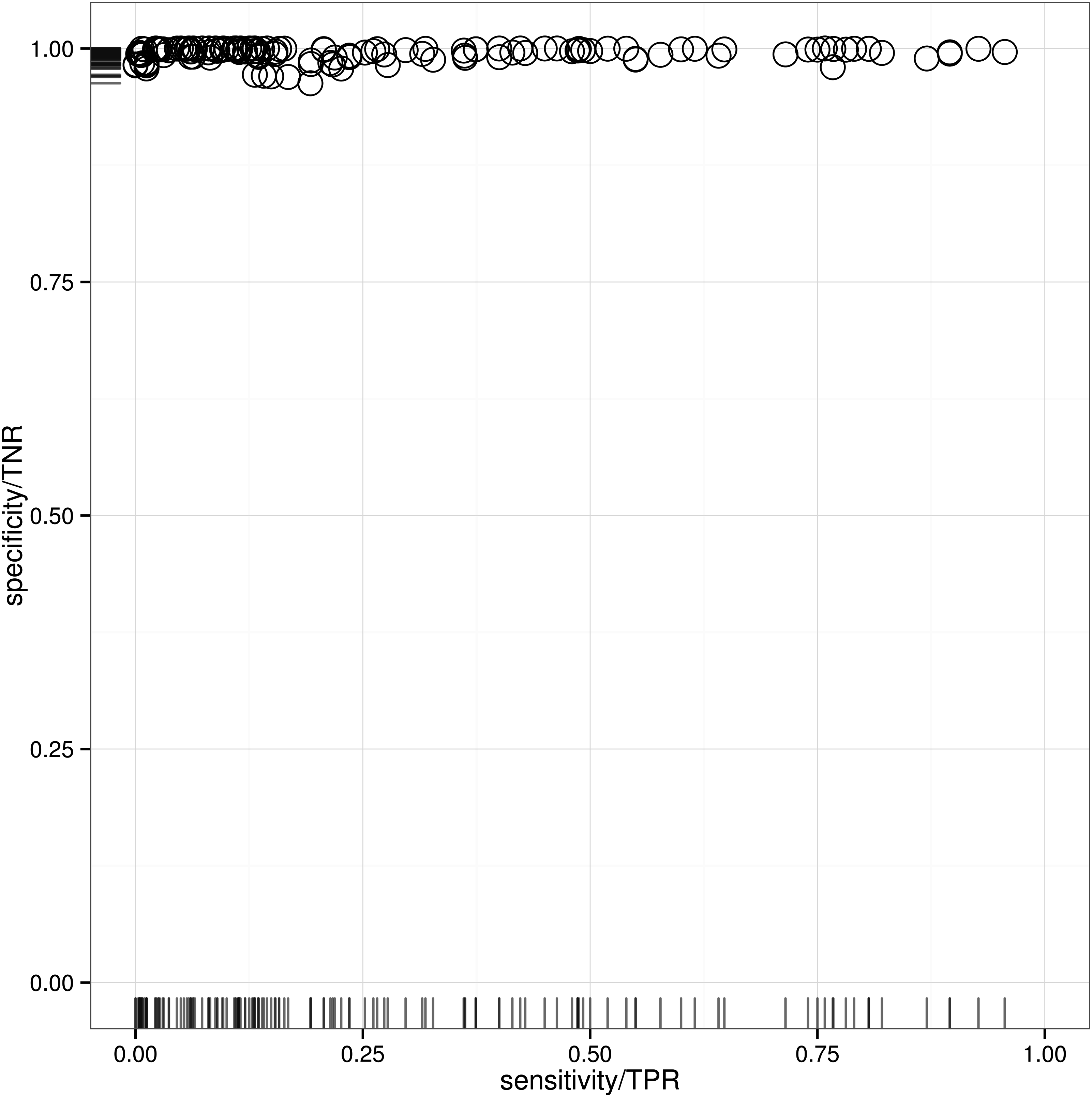
Sensitivity and specificity for Catalytic Site Atlas–derived motifs. Sensitivity or true positive rate (TPR) and specificity or true negative rate (TNR) of the Fit3D search algorithm. Each data point represents a single Catalytic Site Atlas motif; considered matches have p-values ≤0.001. Ideal validation results would be located in the upper right corner of the scatterplot.
3.2. Software
Fit3D was packed to a single software tool programmed in Java in form of an executable JAR file. Furthermore it was purposefully implemented as command line standalone software tool. This application form allows flexibility, automation, and most of all superior integration into problem-specific workflows, especially because of its platform independence thanks to Java. Even extraordinary large datasets, for instance the entire PDB consisting of over 100,000 structures, can be searched conveniently and in an automated way. Primary aspects during development of the Fit3D software were usability and the capability of parameter-free motif matching. This could be realized by the underlying search algorithm, for which the only parameter that may influence results critically is the distance tolerance δ. However, δ is set to a reasonable default value of 1.0 Å, so no care has to be taken in most cases. To run Fit3D the user has to define only two variables: a query motif structure in PDB format and a search target or a list of targets separated by line break. A minimal command line call to start a search for motif KDEEH in a set of target structures can look as simple as the following command:
where m defines the motif structure in PDB format and l a list of target structures in plain text format separated by line break.
The Fit3D software is freely available and extensively documented online. The Supplementary Material (available at www.liebertpub.com/cmb) contains all motif structures mentioned in Table 1 in PDB format. Furthermore, the CSA-derived motifs used for validation are included as well. Additionally, all foreground and background datasets that were used for the validation of the algorithm are included in plain text format. The latest version of the Fit3D implementation is also provided.
4. Conclusions
The mastering of the challenges can be seen as successful: an enhanced algorithm for structural motif matching was developed and validated using gold standard methods. By utilizing multi-atom representation of amino acids it could be shown that sensitivity of our algorithm was superior and nevertheless accompanied with high specificity. Furthermore, algorithmic robustness is guaranteed due to the small number of internal parameters. The novel algorithm was implemented in an easy-to-use command line software tool called Fit3D, which was released under the terms of public licensing and is freely available and ready to be optimized, adapted, or expanded by researchers. Fit3D provides an attractive and improved alternative for existing web services (Nadzirin et al., 2012; Debret et al., 2009; Moll et al., 2011) or standalone tools (Moll et al., 2010). Thereby its application is a double-sided approach: on the one hand it is possible to search for known structural motifs in a large set of target proteins, for example, in the form of a CSA-derived library like the one by Nilmeier et al., (2013). On the other hand, one can screen structures for newly discovered motifs to assess biological function. Due to the rapid growth of automated structure determination methods through structural genomics effort, protein structures are often solved prior to biochemical and functional characterization (Duarte et al., 2012). Hence, our method can be envisioned to be very helpful for researchers dealing with protein crystallography and function determination. The field of drug design and research is also addressed by this approach; the mechanism of drug effect often lies in the inhibition of protein active sites, which are in turn describable through structural motifs. According to the sequence-to-structure-to-function paradigm it can be stated that three-dimensional information is important for the protein chemistry and therefore active sites are conserved (Fetrow and Skolnick, 1998). Additionally, it is evident that ligand binding site resemblance is possible even when global sequence or structure similarity cannot be detected (Xie et al., 2009). It is of high interest for pharmacists to investigate active-site mechanisms and structures in a high-throughput manner. The utilization of search methods for structural patterns can aid researchers to discover off-target binding of specific drugs (Kirshner et al., 2013; Xie et al., 2009). Furthermore, it is conceivable that the approach can be combined with methods for automatic deduction of structural motifs based on functional subfamilies within diverse superfamilies (Redfern et al., 2009). The Fit3D implementation can be seen as a cutting-edge tool in the field of bioinformatic substructure research and computational biology and will be extended and applied in further research.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Acknowledgments
We would like to thank the Free State of Saxony and the Saxon Ministry of Science and Fine Arts for funding this work.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.