
Abstract
Abstract
In studying the strength and specificity of interaction between members of two protein families, key questions center on which pairs of possible partners actually interact, how well they interact, and why they interact while others do not. The advent of large-scale experimental studies of interactions between members of a target family and a diverse set of possible interaction partners offers the opportunity to address these questions. We develop here a method, D
1. Introduction
T
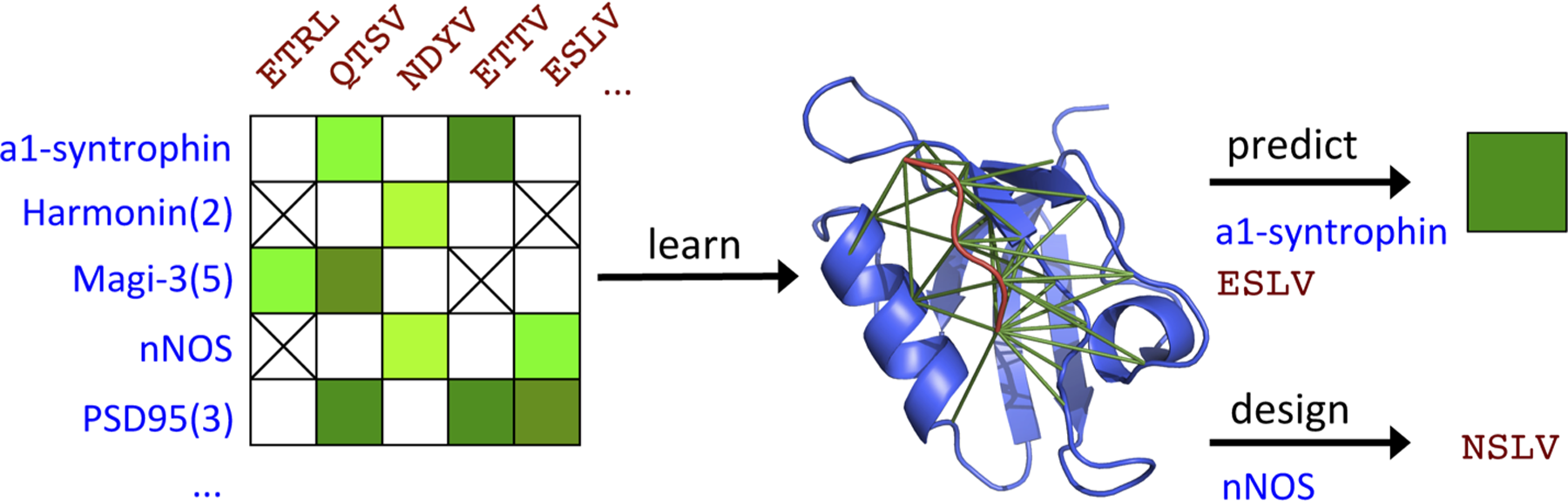
Data-driven Graphical models of Specificity in Protein:protein Interactions (D
As one particular example, consider the specific recognition between PDZ domains and their peptide ligands. PDZs are small peptide recognition modules that bind specific C-terminal peptides of other proteins (Fig. 1, right), in order to mediate protein:protein interactions (e.g., in signaling networks). Early studies of PDZ:peptide recognition developed consensus motifs to capture the common amino acids comprising the ligands of different PDZ “classes” (e.g., class I = S/T-X-Φ vs. class II = Φ-X-Φ, where Φ is a hydrophobic residue). More recent studies yielded more refined statistical binary interaction predictors (interact or not?), based on analysis of amino acid pairs (across the PDZ:peptide interface) in curated datasets of experimentally identified PDZ:ligand partners (Brannetti et al., 2000; Thomas et al., 2009a). MacBeath and colleagues then made the leap to large-scale quantitative data, determining the ΔG of binding for 829 PDZ:peptide pairs from 96 PDZs (from mouse, fly, and worm) against a panel of 259 possible peptide partners in Stiffler et al. (2007). They used this data to develop a binary interaction predictor, based on the constituent PDZ:peptide amino acid pairs like the predictors mentioned above, but taking advantage of the quantitative and negative data in Chen et al. (2008). More recently, Bader and coworkers used the MacBeath data to train a type of support vector regression model for predicting ΔG of binding for PDZ:peptide pairs in Shao et al. (2010).
Motivated by the exciting growth in quantitative studies of protein:protein interactions, we have developed a data-driven, sequence-based model that directly and compactly reveals and represents the amino acid interactions underlying experimentally measured ΔG values of binding (henceforth just ΔG) and enables efficient, accurate, robust, and transparent prediction of ΔGs for new pairs of possible partners (Fig. 1). We employ a graph-structured model (which we refer to simply as a graphical model) that explicitly models amino acid interactions and provides a probabilistic interpretation for them. Sequence-based graphical models of protein families have been used to capture amino acid interactions in order to predict protein structure (Morcos et al., 2011; Jones et al., 2012; Kamisetty et al., 2013) and function (Thomas et al., 2008; Balakrishnan et al., 2011) and design new proteins (Thomas et al., 2009b; Kamisetty et al., 2011b). We build here on our sequence-based models of interacting protein families for binary prediction of interaction developed in Thomas et al. (2009a), significantly extending that approach to incorporate quantitative data and thereby predict ΔG.
We call our approach D
2. Methods
D
2.1. A graphical model of binding free energy
We assume the receptors have been multiply aligned to p informative (nongappy) columns, and the ligands likewise to q residues. Let
Given a receptor sequence
In summary, then, given two protein sequences
2.2. Training objective
Our data-driven approach to modeling protein:protein interaction specificity uses experimental data to learn the parameters V and W that define the model in Equation 1. We assume that the experimental data is partitioned into interactions
We take as our primary objective minimizing the squared error between the observed and predicted ΔG values for members of
Thus our objective function for a specific set of parameters V, W is:
where we emphasize the dependence of ΔGpred in Equation 1 on V and W by including them as parameters. The parameter γ- sets the relative weighting between the contributions from interactions and non-interactions.
2.3. Block-sparse regularization
A suitable model can be learned from the data by minimizing the objective function in Equation 2. However, directly optimizing this function is likely to lead in overfitting as there are usually far more parameters in the model than there are data points available with which to fit them. To circumvent this problem, we instead optimize a regularized objective function. Regularization is usually described as a penalty to the objective function; an alternate but equivalent view of the regularization is that it is a Bayesian prior on the models that biases the learning method toward models consistent with the prior. Protein:protein interactions can be reasonably expected to display structural sparsity—due to spatial restrictions, only a few of all possible bipartite interactions between the partners are likely to be important in biochemical interactions. Motivated by this prior belief, we employ block-L1 regularization, a form of regularization that penalizes the number of nonzero edges (or “blocks”), so that each edge (i, j) is penalized unless all parameters within the edge, Wi,j, are zero. This promotes a sparser structure promoting interpretability; furthermore, by reducing the number of nonzero parameters in the model, it helps avoid overfitting. For our model, the block-L1 regularization term is:
where ∥·∥ refers to the vector two-norm of the corresponding set of parameters and the fraction
Our learning objective is then:
where λ1,2 sets the relative weight between the learning objective and the regularization term.
2.4. Learning algorithms
Schmidt et al. (2009) developed a limited-memory projected quasi-newton (PQN) approach suitable for squared error objectives. We customize their method for our graphical models of protein–protein interactions. A constrained optimization to incorporate the block-L1 regularization is performed by a projected gradient method that iterates between unconstrained gradient descent updates to the parameter values, and constrained projections of the parameter values onto the constrained space.
While this approach can be used to learn the structure and parameters of the model (i.e., which vertices and edges, along with their weights), in practice, the resulting procedure can result in biased weights for the nonzero parameters, despite identifying the correct structure (Meinshausen and Bühlmann, 2006; Balakrishnan et al., 2011). To avoid this, after learning the structure of the model, we relearn the nonzero parameters with L2 regularization. That is, in a second stage, we restrict the optimization to the vertices and edges contributing in the first stage, but reoptimize their weights using a modified version of Equation 4, replacing R1,2 with:
weighted by a corresponding λ2. Since R2(V, W) penalizes the square of the vector 2-norms, each element of each parameter vector is penalized independent of any group membership; the regularization is thus independent of the degrees of freedom in the corresponding groups. This two-stage approach finds sparse models with small edge weights, regularizing a pseudo-likelihood objective similarly to the approach of Balakrishnan et al. (2011). We find in practice that this approach yields models that are both interpretable and predictive of ΔG.
3. Results
Our goal is to make quantitative predictions of the ΔG of PDZ:peptide interactions, interpretable in terms of underlying amino acid constraints. This is in contrast to the approach of Chen et al. (2008), who studied the ability of a computational method to classify interaction vs. noninteraction. [A graphical model approach to do that has been previously described in Thomas et al. (2009a); we have found that classifying based on predicted ΔG is not as robust.] It is also in contrast to the support vector regression approach of Shao et al. (2010), in that while our method achieves comparable predictive accuracy, it has the added benefit of being able to automatically identify the amino acid-level interactions with the greatest impact, and directly characterize their contributions. These interactions not only allow us to characterize the sequence determinants of binding affinity and specificity, but also allow us to design new interacting partners based on the derived “rules” of good interactions.
We apply D
3.1. ΔG prediction
3.1.1. 10-fold cross-validation
To test the ability of our model to predict the affinity of PDZ-peptide interaction, we first performed a ten-fold cross-validation (i.e., we learned the model with 90% of the data and tested it on the left-out 10%, doing this with each 10% left out). This represents the scenario in which data are available for some interactions, and we want to make predictions for others.
Our learning approach has three main parameters: γ−, a parameter trading off the relative importance of positive and negative interactions in the objective function; λ1,2, the strength of the block-L1 regularization used to determine the nonzero parameters of the model, and λ2, the regularization weight used to estimate the values of the nonzero parameters. The γ− and λ2 were set to 0.05 and 1, respectively, based on our initial experiments using one train-test split. The small value of γ− reflects the relative abundance of noninteractions in our dataset and our emphasis on modeling interactions comprehensively since they are biologically more interesting. For each training split, we varied λ1,2 generating multiple models spanning the spectrum from models with no interactions to models where nearly all possible interactions were allowed.
Figure 2 summarizes trends over values of λ1,2. The top panel characterizes the increase in number of interactions with decreasing λ1,2, and the middle panel the corresponding increase in the average Pearson correlation coefficient, from 0.49 when there are no edges in the model and all contributions are due to the Vi, Vj, to 0.66 when ∼60 interactions are included, to a maximum of 0.69 when ∼300 interactions are included. The relatively large increase in model accuracy when the number of edges increases from 0 to 60 suggests that these edges make important contributions to binding affinity and specificity. In contrast, the relatively small increase in accuracy of the model as the number of edges increases beyond 60 to being a completely connected model suggests that the edges introduced later have relatively low importance. The bottom panel shows the average strength of each edge, calculated as the norm of Wi,j, as a function of λ1,2. Each line represents a separate unique interacting pair of residues; interactions that have high weight at λ=25 are highlighted in color, while the remaining interactions are shown in black. When the model is sparse (high λ1,2), there are few, strong interactions; as the density of the model increases (low λ1,2), most interactions have nonzero strength but are very weak.
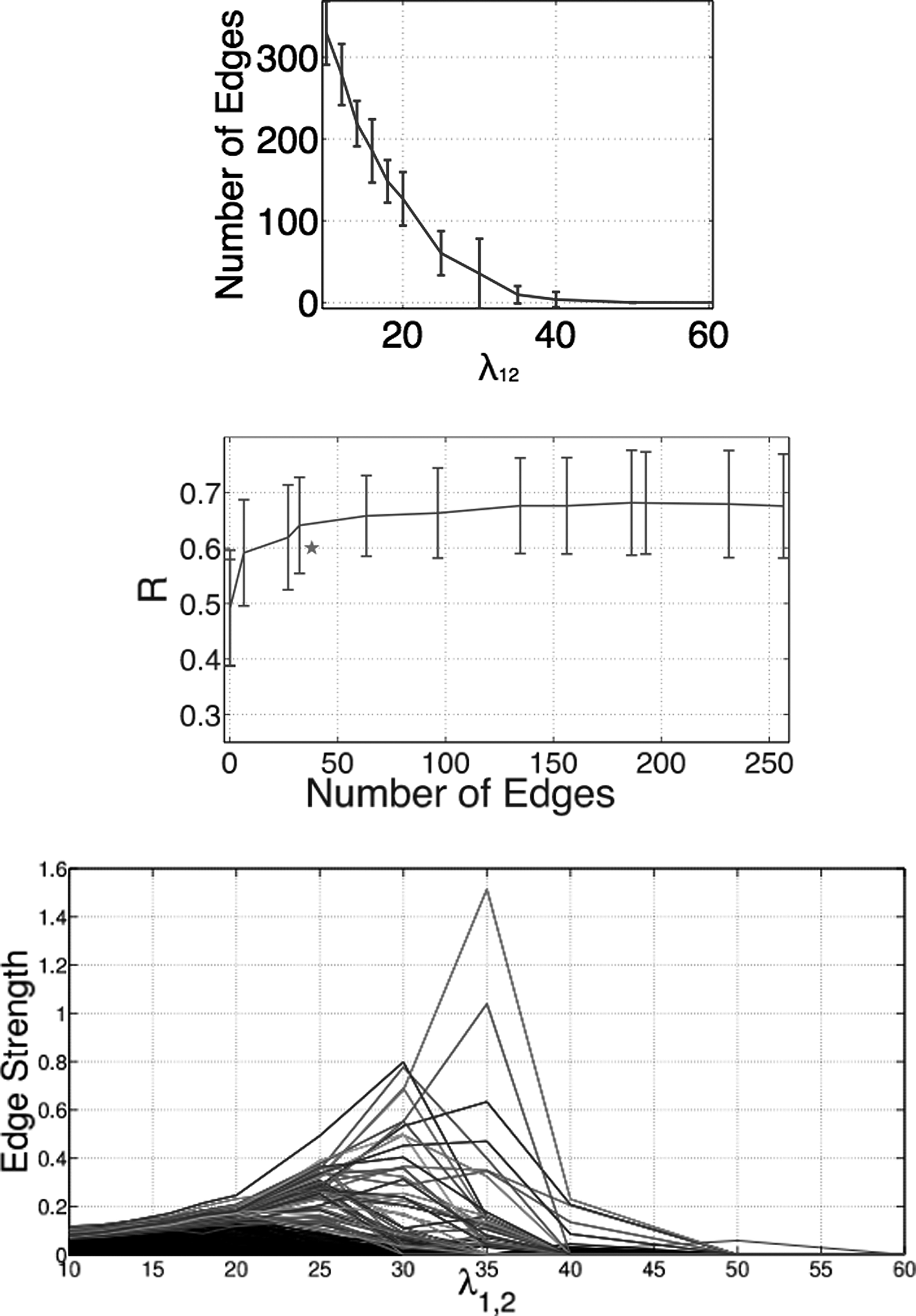
Trends with varying regularization weight (parameter λ1,2), with higher values yielding sparser models. (Top) Number of edges in models learned with varying λ1,2. (Middle) Regularization path, showing each edge's strength as a function of λ1,2. The star indicates a model with edges fixed according to contacts in a crystal structure. (Bottom) Test correlation coefficient as a function of the average number of edges in trained model.
Figure 3 shows the prediction results for one 10-fold repetition at λ1,2=20. The overall correlation coefficient across the dataset was 0.67 while the root mean square error between experiment and prediction was 0.62. Most errors were equally distributed around zero, and actually within typical experimental error. However, there were a few clear outliers where the model under-predicted binding energies.
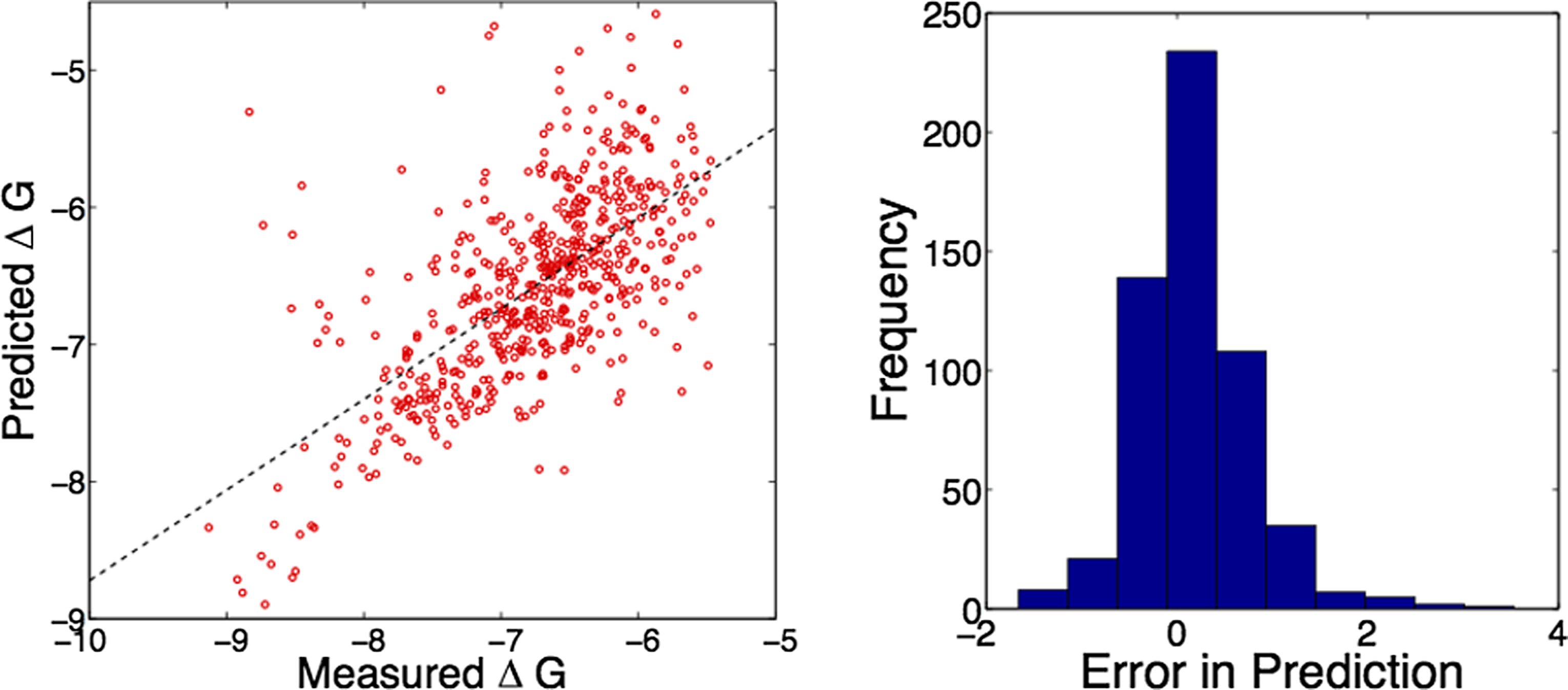
Example prediction results, combined across 10 splits in one repetition at λ1,2 = 20. (Left) Scatterplots of experimental vs. predicted ΔG. Pearson correlation coefficient across entire test-split was 0.67. (Right) Histogram of prediction errors.
3.1.2. Contact-based model structure
When an experimentally determined 3D structure of the protein–protein interface is available, an alternate approach to determining the structure (edges) of the graphical model could be to restrict the nonzero interactions to the pairs of residues close to each other in the 3D structure. The parameters of this model with fixed structure can then be readily learned with L2 regularization, as before. Chen et al. (2008) identified 38 contacts between 16 PDZ residues and 5 peptides. We repeated our 10-fold cross-validation experiments, using these 21 positions and 38 contacting residues as the set of vertices and edges in the model (instead of identifying them using the block-L1 penalty), and estimated their parameters with L2 regularization. The average correlation coefficient of the contact-based models is 0.60, which, while good, is lower than the 0.66 correlation obtained by models with about 60 interactions. Could the difference in accuracy be due to the difference in the number of interactions? The middle panel in Figure 2 highlights the accuracy of this model (shown as a star), compared to the correlation coefficients obtained by varying λ1,2. We see that the models with learned structure can achieve accuracy similar to the contact-structure model but using fewer interactions; alternativeely, a model with learned structure and a comparable number of interactions to that of the contact structure achieves higher correlation. Thus, our data-driven approach to learning model structure can identify important interactions beyond those that might be inferred by inspection of the 3D structure.
3.1.3. Leave-one PDZ out
To test the scenario where the model is applied to make predictions for a new PDZ, we performed “leave-one-PDZ-out” cross-validation following the approach of Shao et al. (2010). We held out data for each of the 23 PDZ domains with at least 10 interactions, training the model on the remaining data and testing on the held-out domain. Since the effect of λ1,2 on the sparsity of the model depends on the number of sequences in the training set, instead of choosing the same value of as selected by ten-fold cross-validation, we performed a grid search on λ1,2 and used the value that gives a model of similar sparsity as the cross-validated models. This process allows us to parameterize the model by the number of edges as opposed to the less natural λ1,2. Using this procedure we obtained an average correlation coefficient of 0.61 across the 23 PDZs that had at least 10 interactions. Again, allowing for denser models by changing the regularization weight slightly improved the average correlation coefficient to 0.63, which is comparable to the 0.65 obtained by Shao et al. (2010) using support vector regression. Table 1 summarizes the correlation coefficient by domain. When restricting to contact edges, we obtain 0.54, about the same as the 0.56 of Bader and colleagues (domain-level details not shown).
Leave-one-PDZ-out cross-validation following Bader and colleagues (Shao et al., 2010) with the published performance of their method (their Table 3) reproduced in the “SemiSVR” column.
3.2. Model analysis
A key feature of D
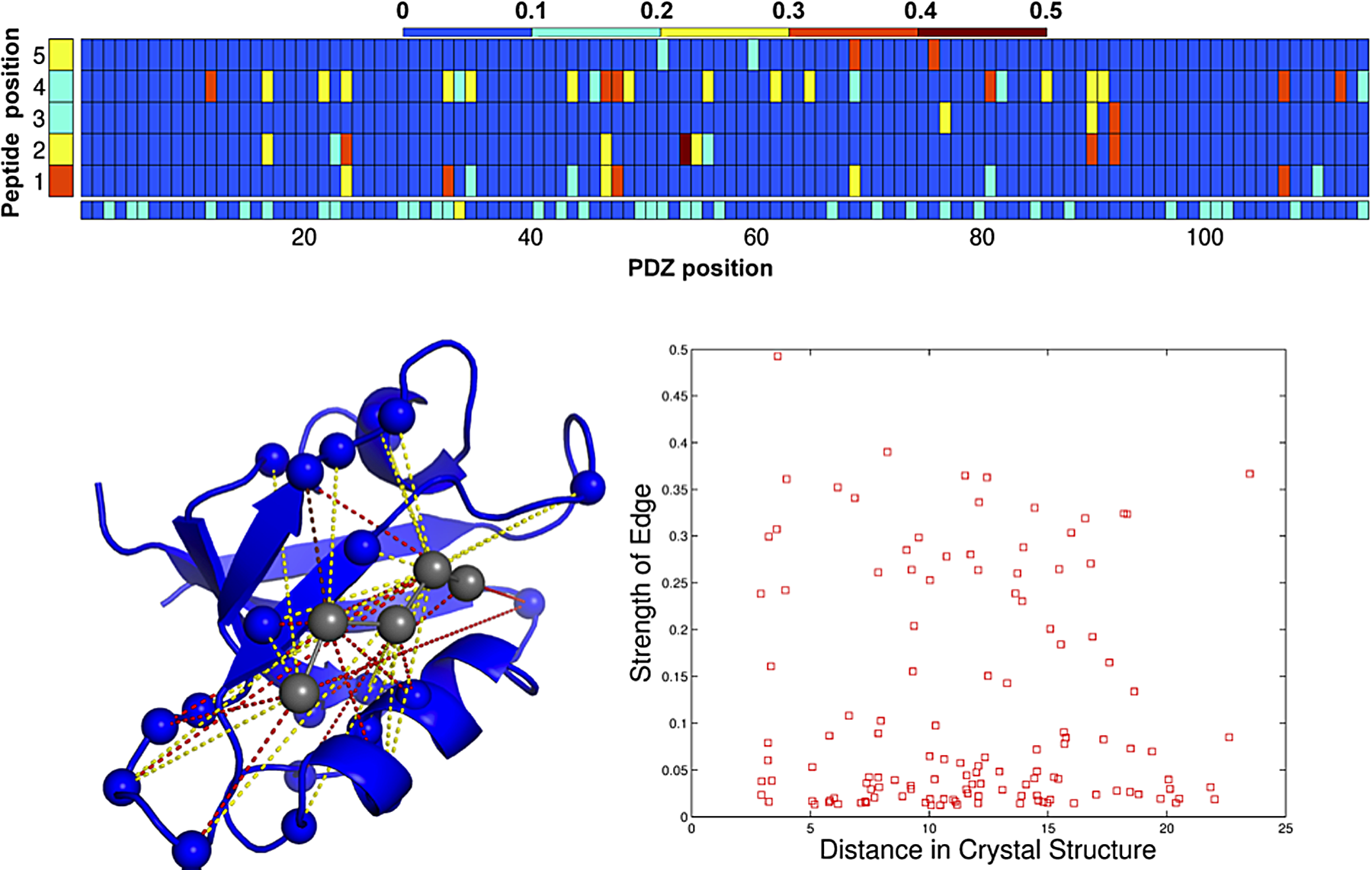
Model analysis. (Top) Average strength of the vector 2-norms for the PDZ positions (i.e., Vi), peptide positions (i.e., Vj), and potentially interacting pairs (i.e., Wi,j) in the model trained at λ1,2=25. (Bottom left) Strong interactions highlighted in top panel, displayed on the NMR structure of the alpha syntropin PDZ (pdb id: 2PDZ). Color scheme same as above. (Bottom right) Average edge strength across 10 training splits plotted against distance in the 3D structure.
Despite the fact that no 3D structure information was used in learning the model, our method identifies several contacting residues as important for determining interaction specificity. This suggests that our data-driven approach might be capturing physically important interactions. To test this hypothesis further, we determined the average weight assigned to each possible amino acid pair for the top three interacting residue pairs across the models for the 10 training folds at λ1,2=25. Figure 5 shows these weights with strong negative energies (i.e., favoring binding) in shades of blue and strong positive energies in shades of red. Darker shades correspond to stronger effects in both cases. The strongest interacting residue pair (PDZ position 54:peptide position 2) strongly favors interactions between oppositely charged arginine/lysine in the PDZ and glutamate in the peptide, while strongly penalizing aspartate/glutamate:glutamate between pairs of negatively charged residues, suggesting a strong electrostatic effect between these positions. Similar effects are seen in the other two interactions with glutamate:lysine favored between 48:1 and aspartate:threonine penalized between 12:4. Our method can thus provide structural information as well as insights into the biochemical determinants of binding affinity.
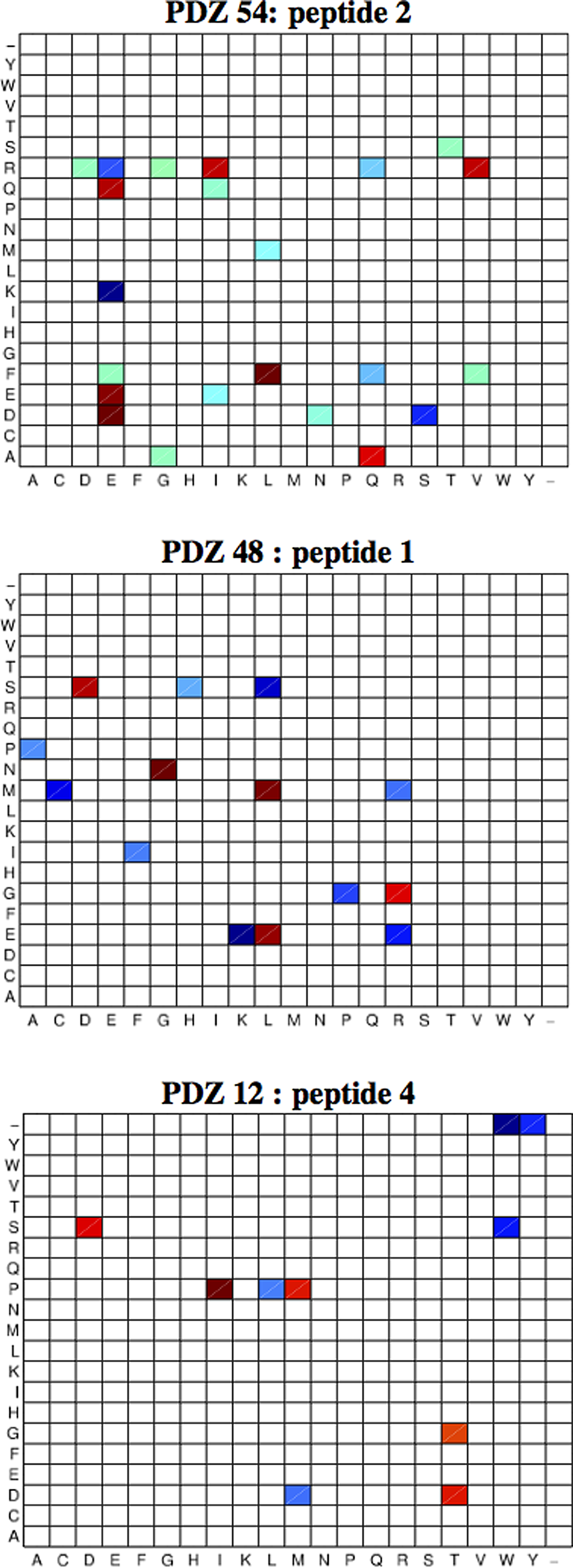
Average weights for amino acid pairs for the top three interacting residue pairs.
In summary, our results suggest that a large fraction of the binding affinity is due to interactions between a relatively small set of positions, not all spatially adjacent to the binding pocket. A larger set of weak interactions might have an additional small effect on binding; these might effect particular subfamilies of PDZs or might reflect allosteric affects related to alternate conformational states of the protein previously described in this family by Lockless and Ranganathan (1999).
3.3. From sequence determinants to sequence design
The accuracy and simplicity of our model allows us to rapidly evaluate the binding affinity of any PDZ–peptide pair. We demonstrate the utility of this approach by “designing” optimal binders for a given PDZ sequence. Using a model learned from the entire training set with λ1,2 = 25, we searched all 5 residue peptides and determined the top 10 peptides by their predicted ΔG for each PDZ sequence. Figure 6 (left) shows the density of predicted binding energies of these PDZ:peptide pairs in blue and that of natural PDZ–peptide pairs binding energies in our training dataset in red. The predicted binding affinities of designed sequences are considerably lower than those of the natural sequences.

(Left) Density of predicted PDZ–peptide ΔG for designed peptides (blue) and experimental ΔG for natural PDZ–peptide pairs (red). (Right) Sequence logos for the top 10 peptide designs for SHANK1, CHAPSYN, and PSD95 (top, middle, and bottom).
While the designed sequences include the natural substrates (at close to their predicted affinities, as discussed above), they also include a diverse array of alternatives. Figure 6 (right) shows sequence logos of the top 10 designed peptides for 3 different PDZs. Even among these sets of top predicted binders, we see interesting diversity among the peptides, suggesting novel designs potentially worthy of experimental evaluation.
4. Discussion and Conclusion
We have developed a graphical model that is highly predictive of the ΔG of binding in protein:protein interactions, while providing an interpretable and designable basis for its predictions. The notion of modularity is fundamental to the idea of a graphical model. Hence these models form a powerful and natural tool to solve problems involving complex probability distributions over many random variables, like the ones here. Due to the natural equivalence between the graph structure of a model and the structure of spatial interactions in proteins, graphical models have seen considerable use in modeling various aspects of proteins: in recognizing structural motifs (Liu et al., 2009; Menke et al., 2010; Moitra et al., 2012), in protein structure alignments (Xu et al., 2006), and in modeling dynamics (Razavian et al., 2012). A growing body of work using graphical models to capture correlated mutations in protein families has also seen substantial success in predicting residue–residue contacts in the protein structure (Marks et al., 2011; Morcos et al., 2011; Nugent and Jones, 2012; Jones et al., 2012; Kamisetty et al., 2013), highlighting the power of these models.
While basing the modeling of ΔG on sequence and data is fundamentally different from structure-based predictors, which employ physics-based models and analysis of side-chain (and potentially backbone) conformations to assess interactions [e.g., Guerois et al. (2002); Kortemme and Baker (2002); Smith and Kortemme (2010)], we note that structure-based undirected graphical models have been used to predict ΔG (Kamisetty et al., 2008, 2011a). The integration of the structure-based approach and the sequence + data-based approach provides an interesting future direction. Our preliminary work on such integration for individual proteins (Kamisetty et al., 2009) provides evidence that the two viewpoints can be complementary and enable better prediction than either alone.
The method we developed here could be applied to any pair of interacting protein families with a similar extent of quantitative binding data. Due to their size and easy availability, PDZ domains form a “model system” for studying protein–protein interactions (Sheng and Sala, 2001; Kurakin et al., 2007; Tonikian et al., 2008; Chen et al., 2008). They are involved in formation of protein complexes that are involved in cellular signal transduction and neural circuitry (Sheng and Sala, 2001) and so make an interesting test case from the point of view of protein engineering (Fuh et al., 2000) and drug design (Saro et al., 2007).
We demonstrated that our models can be used to design novel peptides that interact strongly with a given PDZ domain. This approach could be extended using sampling or other inferential techniques to design a desired interaction, rather than only the peptide, and to scale up to larger sets of involved residues.
Footnotes
Acknowledgments
This work is supported in part by U.S. NSF grant IIS-0905193 (C.J.L. and C.B.K.) and U.S. NIH P41 GM103712 (C.J.L.).
Author Disclosure Statement
The authors declare that no competing financial interests exist.