
Abstract
Abstract
Assembling a genome from short reads currently obtained by next-generation sequencing techniques often results in a collection of contigs, whose relative position and orientation along the genome being sequenced are unknown. Given two sets of contigs, the contig ordering problem is to order and orient the contigs in each set such that the genome rearrangement distance between the resulting sets of ordered and oriented contigs is minimized. In this article, we utilize the permutation groups in algebra to propose a near-linear time algorithm for solving the contig ordering problem under algebraic rearrangement distance, where the algebraic rearrangement distance between two sets of ordered and oriented contigs is the minimum weight of applicable rearrangement operations required to transform one set into the other.
1. Introduction
T
Resequencing is one of the most commonly used approaches in the scaffolding process (Bentley, 2006). The resequencing methods usually require a complete genome of a related organism to serve as a reference. They map the contigs of a draft genome onto a reference genome and then try to infer the ordering and orientation of the contigs according to their positions on the reference genome. Currently, several algorithms based on the resequencing approach have been proposed (van Hijum et al., 2005; Gaul and Blanchette, 2006; Richter et al., 2007; Assefa et al., 2009; Rissman et al., 2009; Muñoz et al., 2010; Husemann and Stoye, 2010; Galardini et al., 2011; Dias et al., 2012; Li et al., 2013; Lu et al., 2014). In fact, all these algorithms can be classified into two categories: alignment-based ones and rearrangement-based ones. The alignment-based algorithms (van Hijum et al., 2005; Richter et al., 2007; Assefa et al., 2009; Rissman et al., 2009; Husemann and Stoye, 2010; Galardini et al., 2011) align contigs or contig ends of a draft genome against a reference sequence, and then ordered and oriented the contigs according to the positions of their matches in the reference. As to the rearrangement-based algorithms (Gaul and Blanchette, 2006; Muñoz et al., 2010; Dias et al., 2012; Li et al., 2013; Lu et al., 2014), they attempt to order and orient the contigs of a draft genome in a way such that the orders of conserved genes (or genetic markers) between the ordered and oriented draft genome and the reference genome are as similar as possible.
When addressing the scaffolding of contigs in a draft genome using the rearrangement-based approach, we formulated the one-sided contig (or block) ordering problem (Li et al., 2013) as follows. Given a draft genome and a reference of complete genome, the one-sided contig ordering problem is to order and orient the contigs of the draft genome such that the genome rearrangement distance between the resulting draft genome and the reference genome is minimized. The sets of ordered and oriented contigs in the resulting draft genome are then called scaffolds. In our previous study (Li et al., 2013), we used the chromosomal algebraic model to represent the draft and reference genomes and utilized the permutation groups in algebra to design an
Recently, Feijão and Meidanis (2013) introduced a new way to model genomes, called the adjacency algebraic model, which can be considered as a combination of the algebraic model we used to solve the one-sided contig ordering problem and the adjacency graph proposed by Bergeron et al. (2006) to simplify the formalization of genomes and the computation of rearrangement distance. The genome rearrangement distance defined under the adjacency algebraic model is called algebraic (rearrangement) distance, which is the minimum weight of applicable rearrangement operations required to transform one of the genomes being compared into the other. Basically, the algebraic rearrangement distance is very similar to, but not the same as, the so-called double-cut-and-join (DCJ) distance (Yancopoulos et al., 2005). The main reason for their difference is due to the different weights assigned to some rearrangement operations. The operations of linear fusion/fission and circularization/linearization have weight ½ in the adjacency algebraic model, while they have weight 1 in the DCJ model. For all other rearrangement operations, such as reversals, transpositions, and block-interchanges, they have the same weight in both models. In this study, we utilize the permutation groups in algebra to propose a near-linear time algorithm for solving the contig ordering problem under the algebraic rearrangement distance, which aims at ordering and orienting the contigs of two input draft genomes such that the algebraic rearrangement distance between the resulting scaffolds of the draft genomes is minimized. Compared with the algorithm proposed by Gaul and Blanchette (2006) based on the breakpoint graphs, our algorithm can be implemented efficiently using simple operations in the permutation groups and the union and find operations of the disjoint-set data structure.
The rest of this article is organized as follows. Some basic concepts and properties of permutation groups in algebra, as well as the algebraic models for representing genomes, are introduced in section 2. In section 3, we present a near-linear time algorithm based on the techniques of the permutation groups to solve the contig ordering problem under the algebraic rearrangement distance. Finally, we make a brief conclusion in section 4.
2. Preliminaries
2.1. Basic concepts of permutation groups
Given a set E={1, 2,…,n}, a permutation α is a one-to-one function from E into itself. Basically, a permutation can be represented by a production of cycles in which each element is followed by its image. For example, α=(1, 3, 2)(4)(5, 6) is a permutation of E={1, 2,…, 6}, meaning that α(1)=3,α(3)=2,α(2)=1, α(4)=4, α(5)=6, and α(6)=5. The elements in a cycle of a permutation can be arranged in any cyclic order. Hence, the cycle (1, 2, 3) of α in the above example can also be rewritten as (2, 3, 1) or (3, 1, 2). If the cycles in a permutation are all disjoint (i.e., no common element in any two cycles), then the product of these cycles is called the cycle decomposition of the permutation. A cycle with k elements is called a k-cycle. Since a 1-cycle represents a fixed element in the permutation, it is usually not written explicitly. For instance, α=(1, 3, 2)(4)(5, 6) can be simply written as α=(1, 3, 2)(5, 6). If the cycles in a permutation are all 1-cycles, then this permutation is called an identity permutation and denoted by
Suppose that α and β are two permutations of E. Then their product αβ, also called composition, defines a permutation of E satisfying αβ(x)=α(β(x)) for all x ∈E. If both α and β are disjoint cycles, then αβ=βα. If αβ=
In fact, every permutation can be expressed into a product of 2-cycles, not necessarily disjoint, in which 1-cycles are still written implicitly. Given a permutation α of E, its norm, denoted by ∥α∥, is defined to be the minimum number, say k, such that α can be expressed as a product of k 2-cycles. In the cycle decomposition of α, let nc(α) denote the number of its disjoint cycles, notably including the 1-cycles not written explicitly. In Huang and Lu (2010), it has been proved that ∥α∥=|E|−nc(α).
Given two permutations α and β of E, α is said to divide β, denoted by α|β, if ∥βα−1∥=∥β∥−∥α∥. The property described below in Lemma 2.1 is useful to determine whether or not a cycle (a1,a2,…,ak) divides the permutation α.
Let α=(a1,a2) be a 2-cycle and β be any permutation of E. Suppose that α|β. Then a1 and a2 are in the same cycle in β according to Lemma 2.1. As a result, this cycle will be broken into two smaller ones in the composition of αβ (or βα). For example, if α=(1, 3) and β=(1, 2, 3, 4), then αβ=(1, 2)(3, 4) and βα=(4, 1)(2, 3). In other words, α functions on β as a split operation. On the other hand, suppose that α ∤β. Then a1 and a2 are in two different cycles in β according to Lemma 2.1. Moreover, these two cycles will be joined into a bigger one in αβ (or βα). For example, if α=(1, 3) and β=(1, 2)(3, 4), then αβ=(1, 2, 3, 4) and βα=(2, 1, 4, 3). Hence, α functions on β as a join operation.
2.2. Genes, chromosomes, and genomes
A gene is an oriented sequence of DNA that starts with a tail and ends with a head. Basically, a gene has two orientations (i.e., forward and backward) and is usually represented by a signed integer, with the sign indicating its orientation, in the studies of genome rearrangements (Lu et al., 2006; Huang and Lu, 2010). A chromosome is then represented by a sequence of ordered genes and a genome by a set of chromosomes. Note that a chromosome can be either linear or circular. In this study, a linear chromosome is further represented as a sequence of ordered genes enclosed in brackets and a circular chromosome as a sequence of ordered genes enclosed in angle brackets. For example, {[1,−2, 3],〈−4, 5,−6〉} denotes a genome consisting of a linear chromosome [1,−2, 3] and a circular chromosome 〈−4, 5,−6〉.
2.3. Representing genomes using a set of adjacencies
For a gene x, its tail and head are also called extremities and denoted by xt and xh, respectively. An adjacency is a set of two extremities to denote a connection between two adjacent genes on a chromosome. A telomere is an extremity not adjacent to any other extremity on a chromosome and represented by a singleton set. Actually, a genome for a given set
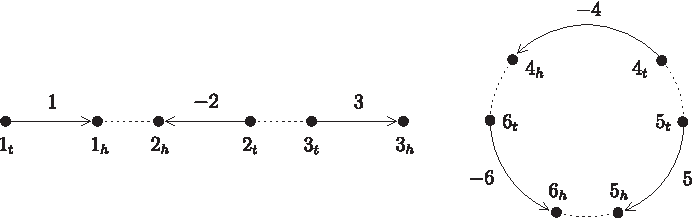
Graph representation of a genome {[1,−2, 3]〈−4, 5,−6〉}, where vertices represent extremities, directed solid edges represent genes, and undirected dotted edges represent adjacencies.
2.4. Representing genomes using algebraic models
Let n be the number of genes and En={−1, 1,−2, 2,…,−n,n} be the set of all genes in forward and backward orientations. The set En is used to model genomes with n genes as permutations. Let Γ=(−1, 1)(−2, 2)…(−n,n) denote the permutation of En that maps each gene into its reverse complement or, equivalently, inverts the sign (orientation) of a gene. It can be easily verified that Γ2=
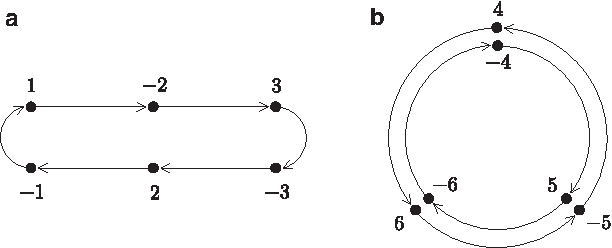
In the chromosomal algebraic model,
The second algebraic model is called adjacency algebraic model, which was introduced by Feijão and Meidanis (2013) to model both circular and linear chromosomes in a uniform way. In the adjacency algebraic model, the tail and head of a gene x are further denoted by +x and −x, respectively (i.e., xt = +x and xh=−x). Moreover, a genome is represented by a permutation that is a product of 2-cycles and 1-cycles, with each 2-cycle corresponding to an adjacency and each 1-cycle corresponding to a telomere in the genome, where the 1-cycles in this permutation can be omitted. For instance, a genome π={[1,−2, 3]〈−4, 5,−6〉} is represented by a permutation πadj=(−1,−2)(2, 3)(4, 5)(−5,−6)(6,−4) in the adjacency algebraic model.
Feijão and Meidanis (2013) proposed an interesting property as follows to show the relationship between chromosomal and adjacency algebraic models. Multiplying a genome on the right by Γ switches the representation of a genome between chromosomal and adjacency algebraic representations. More formally, let πchr and πadj respectively denote the chromosomal and adjacency algebraic representations of a given genome π. Then πchrΓ=πadj and πadjΓ=πchr. For example, suppose that π={[1, −2, 3]〈−4, 5,−6〉}. Then πchr=(1, −2, 3, −3, 2, 1)(−4, 5, −6)(6, −5, 4) and πadj=(−1, −2)(2, 3)(4, 5)(−5, −6)(6,−4). It can be verified that πchrΓ=πadj and πadjΓ=πchr. If ρ is a rearrangement event that can transform πchr into another genome σchr (i.e., ρπchr=σchr), then ρπchrΓ=σchrΓ and hence ρπadj=σadj. It indicates that the rearrangement event acting on a genome can be represented by the same permutation in both the chromosomal and adjacency algebraic models. In addition,
2.5. Algebraic rearrangement problem
As mentioned previously, any set of mutually nonconflicting adjacencies is a valid genome. Therefore, any permutation with a product of disjoint 2-cycles can represent a valid genome in the adjacency algebraic model. Let π and σ be two permutations representing two genomes in the adjacency algebraic representation. A rearrangement operation applicable to a genome π is defined as a permutation ρ that can be applied to π such that ρπ is a valid genome. Moreover, the weight of an applicable rearrangement operation ρ is defined as
According to Lemma 2.2, the norm of σπ−1 is equal to 2d(π,σ). A permutation ρ is said to be a sorting operation from π to σ if ρπ is a valid genome and
By Lemma 2.3, a sorting operation from π to σ is an applicable rearrangement operation on π such that ρ|σπ−1. Feijão and Meidanis (2013) have shown that one can always find a sequence of sorting operations ρ1,ρ2,…,ρk such that ρkρk−1…ρ1π=σ and

The adjacency graph of two genomes π={[1, 4], [3, 2], [−5, 6], [7, 8]} and σ={[1, 2, 3], [4, 5], [6, 7, 8]}, where the vertex corresponding to an adjacency {x,y} is simply labeled as xy and the vertex corresponding to a telomere {z} as z.
3. Algorithm
Note that the genomes considered below are unichromosomal. Given two draft genomes π and σ, with each draft genome represented by a set of contigs, our task in this study is to order and orient the contigs in these two draft genomes such that the algebraic rearrangement distance between the resulting scaffolds of π and σ is minimized. It can be observed that when all the contigs in π and σ are ordered and oriented completely, the adjacency graph obtained from the resulting π and σ has only two paths, if π and σ are two linear chromosomes. This is because only four vertices, which correspond to telomeres in the resulting scaffolds of π and σ, in the adjacency graph have degree one and the others corresponding to adjacencies have degree two. On the other hand, if π and σ are circular chromosomes, then the adjacency graph between the ordered and oriented π and σ has no paths, since all the vertices correspond to adjacencies and thus have degree two. In other words, after completely ordering and orienting all the contigs in both π and σ, the number of paths in their resulting adjacency graph is fixed (either two or zero), meaning that the algebraic rearrangement distance between the ordered and oriented π and σ totally depends on the number of cycles in their adjacency graph according to Theorem 2.1. Therefore, the contig ordering problem under the algebraic rearrangement distance is equivalent to ordering and orienting the contigs in π and σ such that the number of cycles in the adjacency graph between the resulting π and σ is maximized.
Let πchr and σchr denote two draft genomes π and σ respectively in the chromosomal algebraic representation. Note that for a permutation α and an element x, we rewrite α(x) as αx for simplicity, if the context is clear. In fact, ordering and orienting two contigs in πchr or σchr can be considered as a fusion of these two contigs (i.e., joining them into a bigger one). Basically, contigs are linear chromosomal fragments. Then according to the algebraic theory (Feijão and Meidanis, 2013), a fusion of two contigs c1 and c2 in πchr can be modeled by applying a 2-cycle ρ=(Γu,v) to πchr, as illustrated in Figure 4, where Γu and v are telomeres in c1 and c2, respectively. Let πadj and σadj denote π and σ respectively in the adjacency algebraic representation, that is, πadj=πchrΓ and σadj=σchrΓ. In this representation, Γu and v are fixed elements in πadj and hence applying ρ to πadj will join telomeres Γu and v into a 2-cycle (Γu,v). Moreover, such an action will correspondingly lead to telomere vertices Γu and v in the adjacency graph AG(π,σ) being joined as an adjacency vertex {Γu,v}. In AG(π,σ), a telomere vertex must be an end of a path. Suppose that Γu and v are ends in two paths p1 and p2, respectively, in AG(π,σ). Then applying ρ to πadj will also lead to the paths p1 and p2 in AG(π,σ) being joined together as a longer one. However, if p1=p2 (i.e., Γu and v are the ends of the same path), then applying ρ to πadj will cause the path p1 in AG(π,σ) being circularized as a cycle.

Recall that our goal in this study is to find the orderings and orientations of the contigs in two draft genomes π and σ such that the number of cycles in the adjacency graph between the resulting scaffolds of π and σ is maximized. As mentioned above, ordering and orienting two contigs in π or σ is equivalent to finding a fusion to join these two contigs. Moreover, applying a fusion on π or σ to join two of its contigs results in either merging two paths in AG(π,σ) into a longer one or closing a path as a cycle. Hence, the best case occurs when each of all the paths in AG(π,σ) is closed as a cycle by a fusion. Actually, not every path in AG(π,σ) can be closed by a fusion. The criteria for closing a path p in AG(π,σ) by a fusion are as follows: (1) the length of p is even, and (2) both the ends of p correspond to the telomeres in two different contigs in π or σ. If p has odd length, then one end of p corresponds to a telomere of one contig c1 in π and the other end corresponds to a telomere of another contig c2 in σ. In this case, c1 and c2 are in different genomes and hence they cannot be joined together by a fusion. If the length of p is even and both ends of p are the telomeres of the same contig in π or σ, then closing p will create a circular contig, which is not allowed in the process of ordering and orienting contigs. According to the discussion above, we call a fusion as a good fusion if it can close a path in AG(π,σ) when applying to π or σ, and we also call the path being closed as a good path.
In fact, for a nongood path p1 of even length in AG(π,σ), we can join it with any other path p2 by a nongood fusion at any time in the process of ordering and orienting contigs, which does not affect the optimal solution. The reason is described as follows. Basically, there are three cases for p2: (1) p2 is a good path, (2) p2 is an odd path, and (3) p2 is an even but nongood path. If p2 is a good (respectively, odd and nongood even) path, then the join of p1 and p2 through a nongood fusion is still a good (respectively, odd and nongood even) path. It implies that no matter what the path p2 is, the numbers of good and odd paths remain the same after joining p1 with p2, but the number of nongood even paths is decreased by one. Actually, we can imagine that after joining with any other path p2, the nongood even path p1 will disappear without changing anything except extending the length of p2. It suggests that the number of cycles created by joining contigs is not related to the number of nongood even paths. Therefore, it does not matter when we join a nongood even path with another path by using a nongood fusion in the whole process of ordering and orienting contigs.
For a good path p1 in AG(π,σ), it can be joined with any nongood even path, as discussed above. However, it is better to close p1 as a cycle rather than to join it with a good or odd path p2 as a longer path, when we want to find an optimal way to order and orient the contigs in π and σ. The reason is as follows. Suppose that p2 is a good (respectively, odd) path. Then the path obtained by joining p1 and p2 is a good (respectively, odd) path. This indicates that after joining p1 with p2, the number of odd paths, as well as the number of nongood even paths, remains the same, but the number of good paths is decreased by one. This situation is the same as that observed in the case where p1 itself is closed as a cycle. As a result, the greatest number of possible cycles created in the case where p1 is joined with p2 is less than that created in the case where p1 is closed. In other words, closing a good path is better than joining it with another good or odd path, when finding an optimal solution to the contig ordering problem under the algebraic rearrangement distance.
As discussed above, for a good path in AG(π,σ), we can directly close it as a cycle by joining its ends, which corresponds to a good fusion acting on π or σ. However, not all good paths in AG(π,σ) can be closed simultaneously by their corresponding good fusions. It can be observed that closing a good path p1 may cause another good path p2 to become a nongood even one. This may occur when the contigs that can be joined through closing p1 are the same as those that can be joined through closing p2. In this case, closing p1 will lead to both the ends of p2 to become the telomeres of a new contig in π or σ. On the other hand, closing p2 also causes p1 to become a nongood even path. In other words, p1 and p2 cannot be closed simultaneously, suggesting that one of them must be sacrificed. In fact, any of p1 and p2 can be sacrificed.
According to the discussion above, we can first deal with all good paths (by closing them as cycles) until there are no more good paths in the resulting AG(π,σ). After that, the remaining paths must consist of odd paths and nongood even paths. Actually, for those odd paths, we can use the method described below to join them such that the number of the created cycles is maximized. For convenience, we use px,y to denote a path with two ends x and y and, moreover, if the length of px,y is odd, then x and y correspond to the telomeres in π and σ, respectively. First of all, we create an edge-colored graph G=(V,E), called odd path graph, as follows. Each odd path corresponds to a vertex in V. For any two vertices u and v in V, there is a white (respectively, black) edge e∈E between them if there is a contig in current π (respectively, σ) whose both telomeres are two ends of the odd paths corresponding to u and v. For example, consider two draft genomes π={[1, 4], [3, 2], [−5, 6], [7, 8]} and σ={[1, 2, 3], [4, 5], [6, 7, 8]}. There are two good paths in their adjacency graph (Fig. 3). After closing these two paths as two cycles, there are four odd paths—p1t,1t,p4h,6t,p5h,5h, and p8h,8h—as well as one nongood even path p3t,2h, in the resulting adjacency graph. Using these four odd paths, we can create an odd path graph G as shown in Figure 5. Clearly, each vertex in an odd path graph has degree two and hence the odd path graph is a collection of black–white alternating cycles. As mentioned before, an odd path cannot be closed directly as a cycle by a fusion. Hence, at least two odd paths are needed to join them as a cycle. To maximize the number of cycles obtained from the odd paths, the best way is to join every two of them into a cycle by using two fusions. In fact, this is achievable by the following lemma.

An example of an odd path graph, where the dashed and solid lines represent the white and black edges, respectively.
According to Lemma 3.1, we can arbitrarily choose two nonadjacent vertices u and v in the odd path graph G and use two fusions to join their corresponding odd paths, say p1 and p2, into a cycle. These two nonadjacent vertices u and v may come from the same alternating cycle or two different alternating cycles in G. If both u and v are in the same alternating cycle of G, say C, then after joining p1 and p2, either the length of C will be shortened by two, or C will become two smaller alternating cycles C1 and C2 with the sum of their lengths equal to the length of C minus two, that is, |C1| +|C2|=|C|−2 (Fig. 6). If u and v belong to different alternating cycles of G, say C1 and C2, then after the join of p1 and p2, these two alternating cycles will be merged together into a new alternating cycle, say C, whose length is equal to the sum of the lengths of C1 and C2 minus two, that is, |C|=|C1|+|C2|−2 (Fig. 7). In other words, joining two odds paths, which correspond to two nonadjacent vertices in the odd path graph, into a cycle will decrease the sum of the lengths of all the alternating cycles by two. Therefore, we have the following lemma immediately.
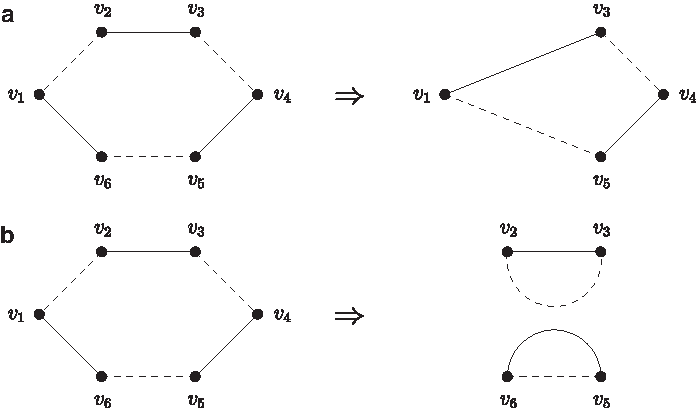
The result obtained by joining two odd paths, which correspond to two nonadjacent vertices u and v in an alternating cycle, into a cycle, where
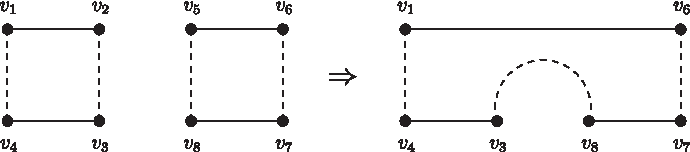
The result obtained by joining two odd paths, which correspond to two nonadjacent vertices u and v in two different alternating cycles, into a cycle, where u=v2 and v=v5.
Based on Lemmas 3.1 and 3.2, we can repeatedly choose any two nonadjacent vertices from the odd path graph and use two fusions to join their corresponding odd paths into a cycle, until the odd path graph becomes an alternating cycle of length two. After that, if there remain nongood even paths in the resulting adjacent graph, then these non-good even paths can be arbitrarily joined with the two odd paths, which correspond to the vertices of the alternating cycle in the final odd path graph, into two longer odd paths. Finally, these two longer odd paths are joined together into a cycle, if the draft genomes π and σ are circular chromosomes.
Our approach described above can be implemented very efficiently using the techniques of the permutation groups in algebra, because Feijão and Meidanis (2013) have demonstrated that there is a direct relationship between the adjacency graph AG(π,σ) and the permutation
According to Lemma 3.3, any cycle of size k in AG(π,σ) corresponds to two disjoint
In fact, the properties mentioned above are helpful for us to use the operations in the permutation groups to determine whether or not a path in AG(π,σ) is a good path, as described in the following lemmas. Given a set X of contigs, let T(X) denote the set of all telomeres in X.
For example, consider the draft genomes π and σ as exemplified previously (also refer to Figure 3 and Table 1). The path p6h,7t in AG(π,σ), which corresponds to the cycle (−6, 7) in σπ−1, is a good path by Lemma 3.7, since −6∈T(π), 7∈T(π) and (−6, 7) ∤ π. However, p3t,2h in AG(π,σ) is not a good path, since in its corresponding cycle (3,−2) in σπ−1, we have 3∈T(π) and−2∈T(π), but (3,−2)|π. Similarly, p3h,4t is a good path in AG(π,σ) according to Lemma 3.8, since it corresponds to the cycle (−3,−1, 4, 2) in σπ−1 with −3∈T(σ), 4∈T(σ) and (−3, 4) ∤σ.
According to the discussion above, we design Algorithm 1 to solve the contig ordering problem for two draft genomes π and σ under the algebraic rearrangement distance. For simplicity, π and σ processed in Algorithm 1 are assumed to be linear. If π and σ are two draft genomes of single circular chromosomes, then the linear contigs returned by Algorithm 1 need to be further circularized into circular contigs by simply joining their telomeres.
In the following, we demonstrate Algorithm 1 by considering two linear draft genomes π={[1, 4], [3, 2], [−5, 6], [7, 8]} and σ={[1, 2, 3], [4, 5], [6, 7, 8]}. By the chromosomal algebraic representation, we have π=(1, 4, −4, −1)(3, 2, −2, −3)(−5, 6, −6, 5)(7, 8, −8, −7) and σ=(1, 2, 3, −3, −2, −1)(4, 5, −5, −4)(6, 7, 8, −8, −7, −6). First of all, we compute σπ−1=(−3, −1, 4, 2)(−4, 5, 6)(3, −2)(−6, 7), in which the cycle (−6, 7) contains two telomeres −6 and 7 from π with (−6, 7) ∤π and the cycle (−3, −1, 4, 2) contains two telomeres −3 and 4 from σ with (−3, 4) ∤σ. Hence, we can apply a good fusion (−6, 7) on π that will join contigs [−5, 6] and [7, 8] into a new one [−5, 6, 7, 8], and apply another good fusion (−3, 4) on σ that will join [1, 2, 3] and [4, 5] into [1, 2, 3, 4, 5]. Actually, applying these two good fusions on π and σ correspond to closing two good paths p6h,7t and p3h,4t in their adjacency graph AG(π,σ) (refer to Fig. 3). After that, we have new σπ−1=(−3, −1)(4, 2)(−4, 5, 6)(3, −2), from which we cannot find any good fusions to apply on current π={[1, 4], [3, 2], [−5, 6, 7, 8]} and σ={[1, 2, 3, 4, 5], [6, 7, 8]}. However, we can see from current σπ−1 that the corresponding adjacency graph AG(π,σ) contains four odd paths p1t,1t,p4h,6t,p5h,5h, and p8h,8h, as well as a nongood even path p3t,2h. The odd path graph created by p1t,1t,p4h,6t,p5h,5h, and p8h,8h is equivalent to that as shown in Figure 5. Next, we choose any one from these four odd paths, say p4h,6t, and find another one p5h,5h that is not adjacent to p4h,6t in the odd path graph (since (−4, −5) ∤π and (6,−5) ∤σ). Hence, we can apply (−4, −5) on π to join two contigs [1, 4] and [−5, 6, 7, 8] into a new one [1, 4, −5, 6, 7, 8] and apply (6, −5) on σ to join [1, 2, 3, 4, 5] and [6, 7, 8] into [1, 2, 3, 4, 5, 6, 7, 8]. This is equivalent to joining the two paths p4h,6t and p5h,5h in current AG(π,σ) into a cycle. We now have new σπ−1=(6, −4)(5,−5)(−3, −1)(4, 2)(3, −2), in which (3, −2) corresponds to the nongood even path p3t,2h in AG(π,σ). Then we choose any one of two remaining odd paths, say p1t,1t, and apply (3, 1) on π, which will join contigs [3, 2] and [1, 4, −5, 6, 7, 8] into a new one [−2, −3, 1, 4, −5, 6, 7, 8]. This action is equivalent to joining p3t,2h and p1t,1t in AG(π,σ) into a longer path p2h,1t. As a result, we finally obtain the scaffolds π={[−2, −3, 1, 4, −5, 6, 7, 8]} and σ={[1, 2, 3, 4, 5, 6, 7, 8]}, and their algebraic rearrangement distance is

In fact, the time complexity of Algorithm 1 mentioned in Theorem 3.1 is near linear in n, since f (n) is a very slowly growing function. Particularly, f (n)≤4 for all practical purposes (Cormen et al., 2009, pp. 561–572), suggesting that the running time of Algorithm 1 can be considered as linear in n in all practical situations.
4. Conclusions
In this work, we studied the contig ordering problem that aims at ordering and orienting the contigs of two input draft genomes such that the genome rearrangement distance between the resulting scaffolds of the draft genomes is minimized. As a result, we presented a near-linear time algorithm to solve this problem under the algebraic rearrangement distance based on the techniques of permutation groups in algebra and the usage of the disjoint set data structure. It is worth mentioning that our algorithm can be done in linear time for all practical situations. The implementation of this algorithm should have a useful application in genome resequencing, especially when only draft genomes (i.e., unfinished genomes) of organisms related to the genome being sequenced are available for reference or comparison.
Author Disclosure Statement
No competing financial interests exist.