
Abstract
Abstract
Ionizing-radiation-resistant bacteria (IRRB) are important in biotechnology. In this context, in silico methods of phenotypic prediction and genotype–phenotype relationship discovery are limited. In this work, we analyzed basal DNA repair proteins of most known proteome sequences of IRRB and ionizing-radiation-sensitive bacteria (IRSB) in order to learn a classifier that correctly predicts this bacterial phenotype. We formulated the problem of predicting bacterial ionizing radiation resistance (IRR) as a multiple-instance learning (MIL) problem, and we proposed a novel approach for this purpose. We provide a MIL-based prediction system that classifies a bacterium to either IRRB or IRSB. The experimental results of the proposed system are satisfactory with 91.5% of successful predictions.
1. Introduction
T
In this work, we study basal DNA repair proteins of IRRB and IRSB to develop a bioinformatics approach for the phenotype prediction of IRR. Thus, we consider that each studied bacterium is represented by a set of DNA repair proteins. Due to this fact, we formalize the problem of predicting IRR in bacteria as an MIL problem in which bacteria represent bags and repair proteins of each bacterium represent instances. Many MIL algorithms have been developed to solve several problems such as predicting types of protein–protein interactions (PPI) (Yamakawa et al., 2007) and drug activity prediction (Fu et al., 2012), mainly including diverse density (Maron and Pérez, 1998), citation-kNN and Bayesian-kNN (Wang and Zucker, 2000), MI-SVM (Andrews et al., 2003), and HyDR-MI (Zafra et al., 2013). Diverse density (DD) was proposed in Maron and Pérez (1998) as a general framework for solving multi-instance learning problems. The main idea of DD approach is to find a concept point in the feature space that are close to at least one instance from every positive bag and meanwhile far away from instances in negative bags. The optimal concept point is defined as the one with the maximum diversity density, which is a measure of how many different positive bags have instances near the point, and how far the negative instances are away from that point. In Wang and Zucker (2000), the minimum Hausdorff distance was used as the bag-level distance metric, defined as the shortest distance between any two instances from each bag. Using this bag-level distance, the k-NN algorithm predicts the label of an unseen bag. In Andrews et al. (2003), the authors proposed the algorithm MI-SVM to modify support vector machines. The algorithm MI-SVM explicitly treats the label instance labels as unobserved hidden variables subject to constraints defined by their bag labels. The goal is to maximize the usual instance margin jointly over the unknown instance labels and a linear or kernelized discriminant function. In Zafra et al. (2013), the authors proposed a feature subset selection method for MIL algorithms called HyDR-MI (hybrid dimensionality reduction method for multiple instance learning). The hybrid consists of the filter component based on an extension of the ReliefF algorithm (Zafra et al., 2012) developed for working with MIL and the wrapper component based on a genetic algorithm that optimizes the search for the best feature subset from a reduced set of features, output by the filter component.
The above cited algorithms use an attribute-value format to represent their data. A most used approach to represent protein sequences in an attribute-value format is to extract motifs that can serve as attributes. Appropriately chosen sequence motifs may reduce noise in the data and indicate active regions of the protein. A protein can then be represented as a set of motifs (Ben-Hur and Brutlag, 2003; Saidi et al., 2012) or as a vector in a vector space spanned by these motifs (Saidi et al., 2010). However, the use of this technique is not suitable in the context of phenotypic prediction of bacterial IRR. This is due to the fact that the set of proteins of each bag must be represented (in the attribute-value format) with the same set of attributes, which is possible only if all extracted motifs from the different bags of proteins were used together as a unique set of motifs. As the different bags of proteins are processed disjointly, it is necessary to design a novel approach for such cases.
In this article, we propose an MIL approach for predicting bacterial IRR using proteins implicated in basal DNA repair. For this purpose, we used a local alignment technique to measure the similarity between protein sequences of the studied bacteria. To the best of our knowledge, this is the first work that proposes an in silico approach for phenotypic prediction of bacterial IRR.
The remainder of this article is organized as follows. Section 2 presents the materials and methods used in our study. In section 3, we describe our experimental techniques and we discuss the obtained results. Concluding points make the body of section 4.
2. Materials and Methods
2.1. Terminology and problem formulation
The task of multiple instance learning (MIL) was coined by Dietterich et al. (1997) when they were investigating the problem of drug activity prediction. In multiple-instance learning, the training set is composed of n labeled bags. Each bag in the training set contains k instances and have a bag label yi ∈{–1, +1}. We notice that instances of each bag have labels yij ∈{–1, +1}, but these values are not known during training. The most common assumption in this field is that a bag is labeled positive if at least one of its instances is positive, which can be expressed as follows:
The task of MIL is to learn a classifier from the training set that correctly predicts unseen bags. Although MIL is quite similar to traditional supervised learning, the main difference between the two approaches can be found in the class labels provided by the data. According to the specification given by Dietterich et al. (1997), in a traditional setting of machine learning, an object m is represented by a feature vector (an instance), which is associated with a label. However, in a multiple instance setting, each object m may have k various instances denoted
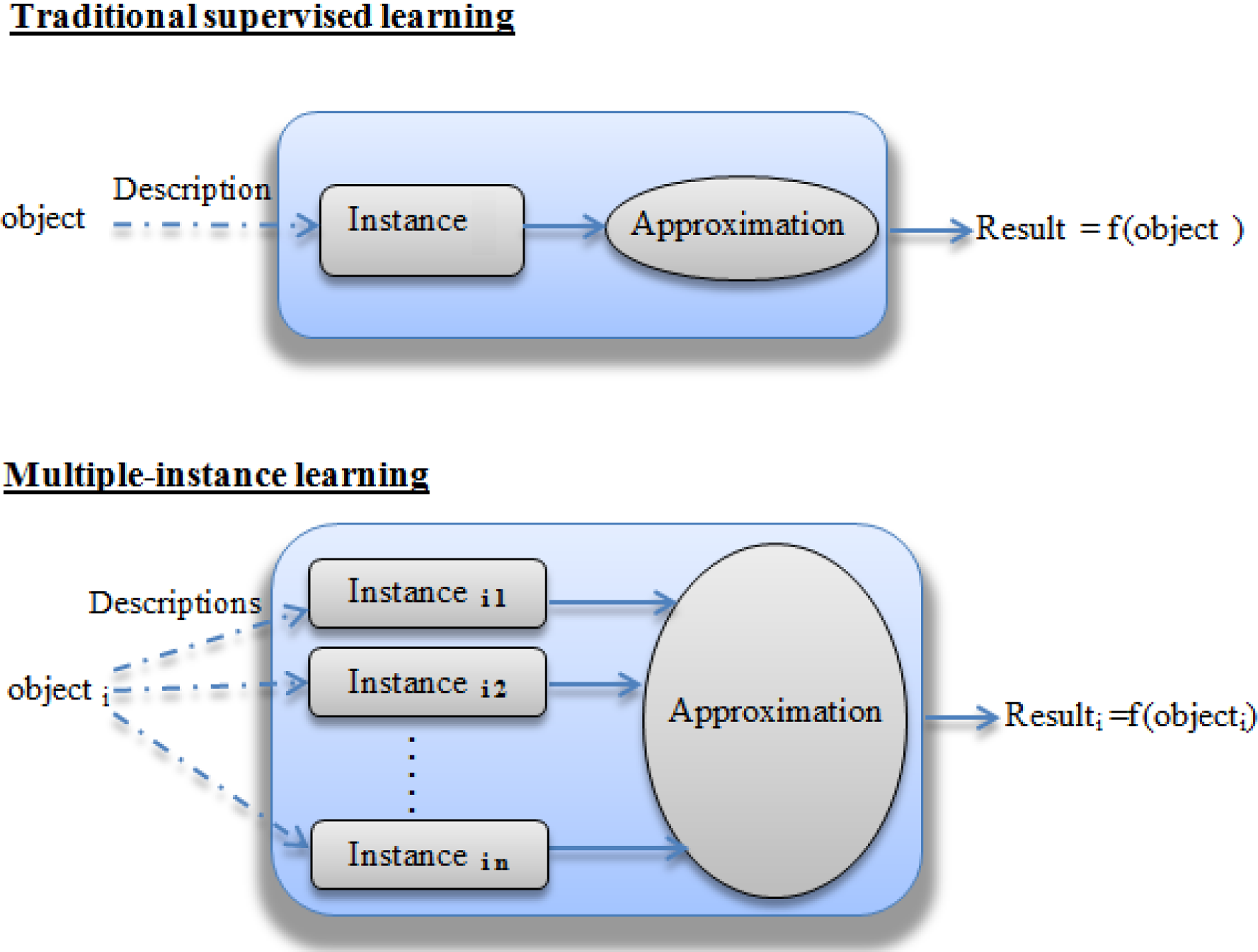
Differences between traditional supervised learning and multiple instance learning.
In our work, we are interested in the prediction of the phenotype of IRR in a family composed of a set of bacteria. Let
The problem investigated in this work is to learn a multiple-instance classifier in this setting. Given a query bacterium
2.2. MIL-ALIGN algorithm
Based on the formalization, we propose the MIL-ALIGN algorithm allowing to predict IRRB. The proposed algorithm focuses on discriminating bags by the use of local alignment technique to measure the similarity between each protein sequence in the query bag and corresponding protein sequence in the different bags of the learning database.
In MIL-ALIGN algorithm we use the following variables for input data and for accumulating data during the execution of the algorithm:
• the variable Q: corresponds to the query bag (the query bacterium), which is a vector of protein sequences. • the variable DB: corresponds to the bacteria database. • the variable S: corresponds to a matrix used to store alignment score vectors.
Informally, the algorithm works as follows (see Algorithm 1):
1. For each protein sequence pi in the query bag Q, MIL-ALIGN computes the corresponding alignment scores (line 1 to 5). 2. Group alignment scores of all protein sequences of query bacterium into a matrix S (line 3). Line i of S corresponds to a score vector of protein pi against all proteins 3. Apply an aggregation method to S in order to compute the final prediction result R (line 6 to 7). A query bacterium is predicted as IRRB (respectively IRSB) if the aggregation result of similarity scores of its proteins against associated proteins in the learning database is IRRB (respectively IRSB).
2.3. Experimental environment
Information on complete and ongoing IRRB genome sequencing projects was obtained from the GOLD database (Liolios et al., 2008). We initiated our analyses by retrieving orthologous proteins implicated in basal DNA repair in IRRB and IRSB with sequenced genomes. Proteins of the bacterium Deinococcus radiodurans (B7) were downloaded from the UniProt website. PrfectBLAST tool (Santiago-Sotelo and Ramirez-Prado, 2012) was used to identify orthologous proteins. Proteomes of other bacteria were downloaded from the NCBI FTP website.
For our experiments, we constructed a database containing 28 bags (14 IRRB and 14 IRSB). Table 1 presents the used IRRB and IRSB. Each bacterium contains 25 to 31 instances that correspond to proteins implicated in basal DNA repair in IRRB (see Table 2).
D10: Dose for 90% reduction in colony forming units (CFUs); for IRRB, it is greater than 1 kGy.
Chroococcidiopsis spp.
Kocuria rosea.
T. thermophilus HB27.
IRRB, ionizing-radiation-resistant bacteria; IRSB, ionizing-radiation-sensitive bacteria.
3. Results and Discussion
3.1. Experimental techniques
Computations were carried out on an i7 CPU 2.49 GHz PC with 6 GB memory, operating on Linux Ubuntu. In the classification process, we used the leave-one-out (LOO) technique (Han et al., 2011) also known as jack-knife test. For each dataset (comprising n bags), only one bag is kept for the test and the remaining part is used for the training. This action is repeated n times. In our context, the leave-one-out is considered to be the most objective test technique compared to the other ones (i.e., hold-out, n-cross-validation), as our training set contains a small number of bacteria.
For our tests, we used the basic local alignment search tool (BLAST) (Altschul et al., 1990) for computing local alignments. We implemented two aggregation methods to be used with MIL-ALIGN: the sum of maximum scores method and the weighted average of maximum scores method.
Below, we formally define the SMS method:
where
•
•
Below, we formally define the WAMS method:
where
•
•
and
•
•
where wi is the weight of the protein pi.
3.2. Results
In order to simulate the traditional setting of machine learning in the context of prediction of IRR in bacteria, we conducted a set of experiments with MIL-ALIGN by selecting just one protein for each bacterium in the learning set. Each experiment consists of aggregating alignment scores between a protein sequence of a query bacterium and the corresponding protein sequences of each bacterium in the learning database. We present in Table 3 learning results with the traditional setting of machine learning. The LOO-based evaluation technique was used to generate the presented results.
As shown in Table 3, we conducted 31 experiments (with 31 proteins). Results show that the use of our algorithm with just one instance for each bag in the learning database allows good accuracy values.
In order to study the importance of considering the problem of predicting bacterial IRR as a multiple instance learning problem, we present in Table 4 the experimental results of MIL-ALIGN using a set of proteins to represent the studied bacteria. For each set of proteins and for each aggregation method, we present the accuracy, the sensitivity, and the specificity of MIL-ALIGN. We notice that the WAMS aggregation method was used with equally weighted proteins. We used the LOO-based evaluation technique to generate the presented results.
SMS, sum of maximum scores; WAMS, weighted average of maximum scores.
We notice that the use of the whole set of proteins to represent the studied bacteria allows good accuracy accompanied by high values of sensitivity and specificity. This can be explained by the pertinent choice of basal DNA repair proteins to predict the phenotype of IRR. The high values of specificity presented by MIL-ALIGN indicate the ability of this algorithm to identify negative bags (IRSB). Using all proteins, we have 92.8% accuracy and specificity. We do not exceed these values in all the cases of mono-instance learning presented in Table 3. As shown in Table 4, the SMS aggregation method allows better results than the WAMS aggregation method using the whole set of proteins to represent the studied bacteria. Using the other subsets of proteins (DNA polymerase, replication complex, and other DNA-associated proteins) to represent the bacteria, SMS and WAMS present the same results.
In order to study the correctly classified bacteria with the MIL, we computed for each bacterium in the learning database the percentage of experiments that succeed to classify the bacterium (see Table 5).
Successfully classified bacterium using three settings: (1) all proteins with SMS aggregation method; (2) replication complex proteins with SMS and WAMS aggregation methods; and (3) other DNA-associated proteins with SMS and WAMS aggregation methods.
As shown in Table 5, more than 89% of tested bacteria show successful predictions of 100%. This means that we succeed to correctly predict the IRR phenotype of those bacteria. On the other hand, the results illustrated in Table 5 may help to understand some characteristics of the studied bacteria. In particular, the IRRB M. radiotolerans (B11) and the IRSB B. abortus (B15) present a high rate of failed predictions. It means that in most cases, M. radiotolerans is predicted as IRSB and B. abortus is predicted as IRRB; the former is an intracellular parasite (Halling et al., 2005) and the latter is an endosymbiont of most plant species (Fedorov et al., 2013). A probable explanation for these two failed predictions is the increased rate of sequence evolution in endosymbiotic bacteria (Woolfit and Bromham, 2003). As our training set is composed mainly of members of the phylum Deinococcus-Thermus; expectedly, the Deinococcus bacteria (B2-B7) present a very low rate of failed predictions.
4. Conclusion
In this article, we addressed the issue of prediction of bacterial IRR phenotype. We have considered that this problem is a multiple-instance learning problem in which bacteria represent bags and repair proteins of each bacterium represent instances. We have formulated the studied problem and described our proposed algorithm MIL-ALIGN for phenotype prediction in the case of IRRB. By running experiments on a real dataset, we have shown that experimental results of MIL-ALIGN are satisfactory with 91.5% of successful predictions.
In the future work, we will study the performance of the proposed approach to improve its efficiency, particularly for endosymbiont bacteria. Also, we will study the use of a priori knowledge to improve the efficiency of our algorithm. This a priori knowledge can be used to assign weights to proteins during the learning step of our approach. A notable interest will be dedicated to the study of other proteins that can be involved with the high resistance of IRRB to the IR and desiccation, two positively correlated phenotypes.
Footnotes
Acknowledgments
This work was partially supported by the French-Tunisian project: General Directorate of Scientific Research in Tunisia (DGRST)/National Center for Scientific Research in France (CNRS) [IRRB11/R-14-09], by the French Region of Auvergne, and by the Fédération de Recherche en Environment [UBP/CNRS-FR-3467].
Author Disclosure Statement
The authors declare they have no conflicting financial interests.