
Abstract
Abstract
The p53 protein is a stress response protein that functions primarily as a tetrameric transcription factor. A tumor suppressor p53 binds to a specific DNA sequence and transactivates target genes, leading to cell cycle apoptosis. Encoded by the human gene TP53, p53 is a stress response protein that functions primarily as a tetrameric transcription factor. This gene regulates a large number of genes in response to a variety of cellular functions, including oncogene activation and DNA damage. Mutations in p53 are common in human cancer types. Herein we mutate a wild-type p53, 1TSR with four of its mutated proteins. The energy for the wild-type and mutated proteins is calculated by using molecular dynamics simulations along with simulated annealing. Our results show significant differences in energy between hotspot mutations and the wild type. Based on the findings, we investigate the correlation between molar masses of the target residue and the relative energy with respect to the wild type. Our results indicate that the relative energy changes play a pivotal role in bioactivity, in conformity with observations in the rate of mutation in biology.
1. Introduction
S
In addition, p53, stabilized in response to cellular stress such as DNA damage and oncogene activation, binds specific DNA targets as a tetramer and regulates the expression of genes involved in cell cycle arrest, DNA repair, or apoptosis (Vogelstein et al., 2000; Ho et al., 2006). The p53 has a modular structure with multiple domains, including a core DNA-binding (residues ∼94–292) and a tetramerization domain (residues 325–356) (Ho et al., 2006; Kitayner et al., 2006; Ma et al., 2007). Both domains are connected by a flexible linker (residues 293–324), are structurally folded, and manifest a well-defined three-dimensional structure as compared to the N- and C-terminal regions (residues 1–93 and 357–393, respectively), which are largely unstructured (Ho et al., 2006; Kitayner et al., 2006; Joerger and Fersht, 2008). These unstructured regions harbor the transactivation domain and motifs that can modulate the function of p53 through specific posttranslational modifications and interactions with other proteins (Bell et al., 2002; Gu and Zhu, 2012).
More than 80% of missense mutations in TP53 found in human cancers are located in the core DNA domain (Hollstein et al., 1991; Olivier et al., 2002). Several of these mutations disrupt DNA binding directly or reduce the folding/stability of the core domain, suggesting that DNA binding is critical to p53 function.
Studies of the core domain of wild-type p53 have revealed the details of protein folding and DNA recognition, and given insight into potential mechanisms of cancer-associated mutations. Some of these missense mutations are temperature sensitive; for example, P223L and V274F in DU145 prostate cancer cells retain transactivation functions at lower temperatures (32°C) (Bajgelman and Strauss, 2006), while other mutations result in complete loss of activity (gain of function/loss of transactivation). Structural studies suggest that the temperature-sensitive mutants and other mutants promote near wild-type structure of p53 under certain conditions. The protein–protein interaction between wild-type and mutant p53 core domains in higher-order p53 complexes has also been a subject of experimental and modeling studies (Klein et al., 2001; Rippin et al., 2002; Ho et al., 2006; Kitayner et al., 2006; Ma et al., 2007; Chen et al., 2010). Based on these results, we hypothesized that theoretical modeling of p53 and its mutants could provide a parameter that can link the type of mutation with p53 structure and possibly function. These structural simulation parameters of various known p53 mutations could be used to establish an association between cancer prognosis, response to treatment, and more importantly develop strategies to recover the structure of mutant p53 to simulate as a wild-type protein. Studies have suggested that common p53 cancer mutants exhibit a variety of distinct local structural changes, while the overall structural scaffold remains largely preserved (Joerger and Fersht, 2008). Thus, a focus on the structural changes associated with the local core DNA-binding domain may be more informative in terms associating the severity of mutants with function.
Modeling via molecular docking, molecular dynamics (MD) simulation, and free energy prediction of binding are routinely applied to supplement the experimental studies in order to understand the underlying mechanism at an atomic level for improving the efficiency of molecular design or discovery (Fu et al., 2012). Theoretical modeling of p53, and conformation changes induced by protein–protein interactions, as well as peptide and small molecular inhibitors have significantly expanded our current understanding of the dynamic nature of p53 structure (Hollstein et al., 1991; Fu et al., 2012). While there have been several studies discussing the dynamic simulations of p53 in recent years, the dynamics and flexibility of mutation-induced conformational changes of p53 have not been completely characterized or understood. Moreover, the dynamic characteristic of other domains of p53 has not been fully studied. In this study, we aim to understand via computational analysis of p53 structural models the energy differences of wild type and mutant forms of p53. Moreover, this study will aid in providing better understanding of how disparate energy, temperature sensitivity, and missense mutation differences among different forms of mutant p53 compared to wild type affect the disease state of cancer.
2. Methods
2.1. The structures of wild-type and mutant p53
The wild-type p53 core domain (residues 94–269) structure 1TSR (Cho et al., 1994) (Fig. 1) was obtained from the Protein Data Bank (PDB), a widely used portal for biological macromolecular structures (Berman et al., 2003). The 1TSR represents the X-ray crystal structure of a wild-type core domain p53-DNA complex at 2.2 Å resolution with a crystallographic R factor of 20.5% (Cho et al., 1994). The DNA binding domain of p53 contains a beta sandwich that serves as a scaffold for two large loops and a loop-sheet-helix motif. The two loops, which are held together in part by a tetrahedrally coordinated zinc atom, along with the loop-sheet-helix motif, form the DNA binding surface of p53. Therefore, we used 1TSR as a framework to study p53 mutations that fall within the DNA binding domain. The base 1TSR peptide sequence was used to introduce prototype p53 mutations: R175H, R273H, P223L, and V274F. The widespread R175H and R273H were used as hotspot gain-of-function p53 mutations, while P223L and V274F were used as weak temperature-sensitive mutants found in DU145 prostate cancer cell lines.
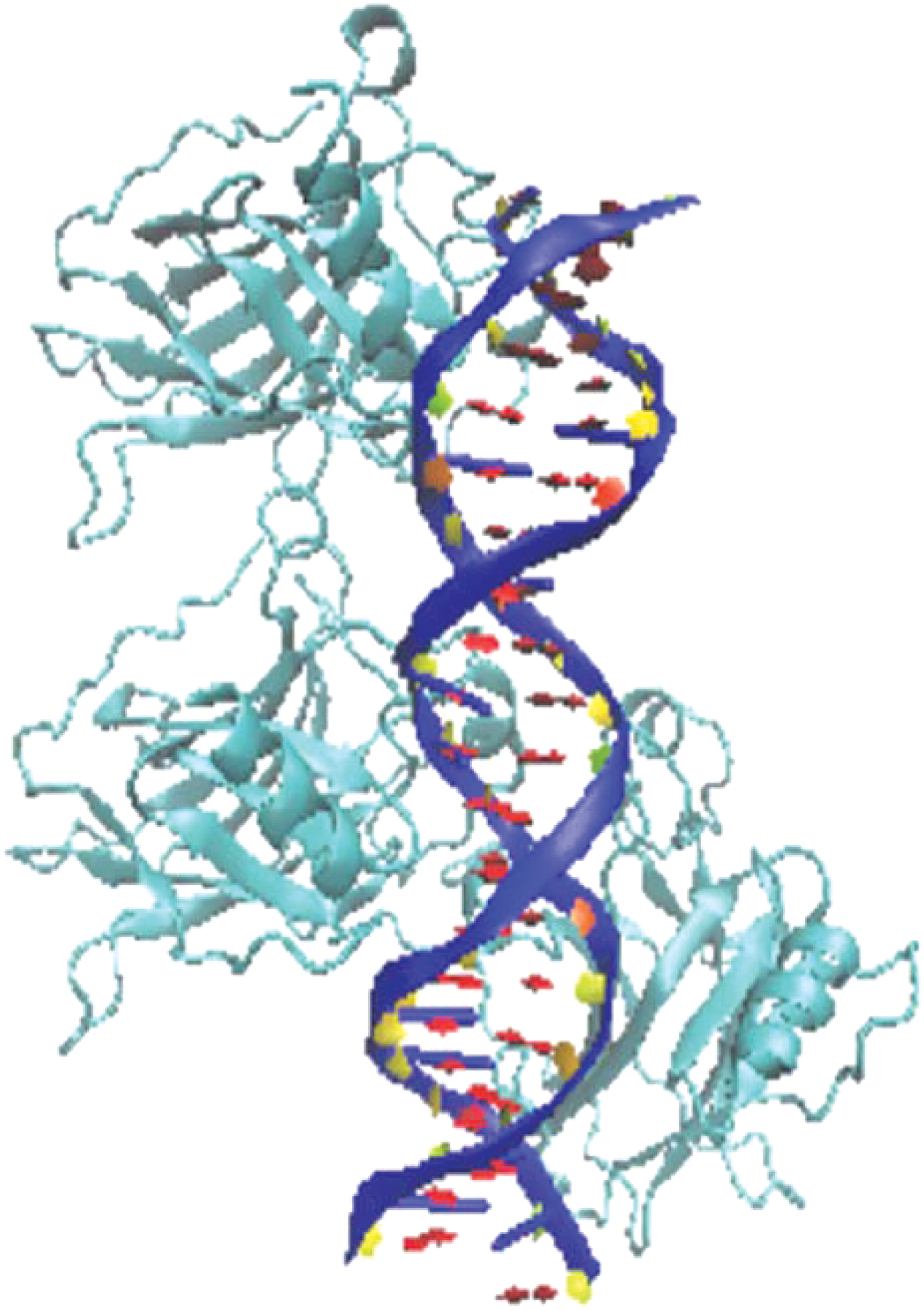
1TSR. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations.
2.2. Molecular dynamics simulation
We performed molecular dynamic simulations of 1TSR (wild type) and core domain mutants using the CHARMM (Chemistry at HARvard Macromolecular Mechanics), an all-atom empirical energy function force field (Brooks et al., 1983, 2009; MacKerell et al., 1998). The CHARMM force field treats interactions of amino acids with bonded and nonbonded terms. It is worth noting that, to properly characterize the relevant interactions, it is necessary to include an explicit solvent. For this reason, our MD calculation results were rectified with simulations with explicit water (Phillips et al., 2005).
Computational simulations largely utilize the atomic description of biomolecules. However, many studies have shown that molecular systems may have diverse conformations because of large numbers of degrees of freedom around chemical bonds, leading to a plethora of local minima conformations (Adcock and McCammon, 2006). Biological function is associated when the structure is in the proximity of a global energy minimum. In an effort to optimize the system, we used simulated annealing with repeated search cycles with various initial conformations using nanoscale molecular dynamics (NAMD) and subsequent visualization in visual molecular dynamics. NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign (Phillips et al., 2005). NAMD is a parallel molecular dynamics platform (on Charm++) for high-performance simulation of large biomolecules systems. Simulated annealing serves as an optimization technique that the energy of biomolecules readily converges after 20–30 cycles.
The potential energy of core domain 1TSR and mutants was calculated in a given conformation as a sum of individual energy terms of bonded and nonbonded interactions (MacKerell et al., 1998; Phillips et al., 2005; Brooks et al., 2009). The following assumptions were made to address the effect of mutation in a confirmation: (1) changing a residue in a wild type affects overall bonding; (2) nonbonded terms such as electrostatic, van der Waals, and polarization are independent of mass; and (3) bonded terms such as energy of stretch, bending, and torsion depend on the molar mass of residues. A bond was considered as a spring with its equilibrium length r0 (in our assumption the center of mass of a residue is r0). The energy that is required to stretch or compress a bond between two atoms or to bend a bond from its equilibrium angle depends on the constant of the spring constant. Therefore, the overall energy of bending and stretching depends on molar mass while a mutation is considered similar to changing the spring in a harmonic motion. Periodic Fourier function was used to represent the energy of torsions between particles based on molecular dynamics. The interaction mode vn was defined as
We also assumed at the minimum that mutating a residue in a protein may have an effect on at least two near-neighbor residues (Brocchieri and Karlin, 1995; Deutsch and Krishnamoorthy, 2007). The molar mass was therefore extracted from the PDB file. Specifically, for the residues involved, the effective mass is calculated as
where the nominator and denominator denote the reciprocal effective mass for the mutated residue and wild type, respectively.
Finally, we consider the effective mass of a three-sequence segment as
where the central segment can be either a mutated or wild-type residue. As such, we can characterize the relative change of the effective mass as
Thus, μ is dimensionless and characterizes the correlation between the mutated protein and the wild-type protein.
3. Results and Discussion
The study was inspired by experimental observations that focused on the restoration of mutant p53 because of the intrinsic flexibility of its domain structure (Bell et al., 2002). Certain p53 mutants can be rescued via temperature (temperature-sensitive mutants p223L and V274F), and artificial means such as small molecules and short peptides (reviewed in Li et al., 2015). Our studies also revealed that mutant p53 could be restored to function as wild-type via physiological means through acetylation of lysine residues (K373) (Knowell et al., 2013). Thermodynamic studies revealed that the R248Q mutant is stable at subphysiological low temperatures (Bullock et al., 1997). The R248Q stability was less than that of the wild-type protein by ∼2 kcal/mol. The mutant structure also engaged in a two-stage unfolding transition, analogous to the wild-type protein (Ishimaru et al., 2003), which indicated well-defined structures at low temperature.
The p53-reactivation and induction of massive apoptosis-1 (PRIMA-1) (Bykov et al., 2002) and its methylated form, RRIMA-1MET (APR-246) (Bykov et al., 2005), were shown to restore sequence-specific DNA binding region of R273H and R175H by forming adducts with thiols in mutant p53 and activating several p53 target genes and promoting apoptosis in human cancer cells (Lambert et al., 2009). Thus, thermodynamic stability calculated through global energy minimization simulations provided insight into the structural flexibility of mutant p53. Our long-term goal is to use these simulations to discover mutants that could be (1) sensitive to restoration of mutant p53 biological activity and (2) severity of the mutant that can subsequently be used to predict prognosis.
The calculated energies based on the simulated annealing of the wild-type and mutated core domain of p53 are shown in Table 1 and Figure 2A. The total calculated energies (E) and normalized calculated energies per atom (Ea) were lower for wild-type p53 (−19244 and −2.123 kcal/mol, respectively) as compared to the normalized energies per atom of hotspot mutants R175H and R273H (−2.0 kcal/mol), suggesting that the wild-type p53 core domain was more thermodynamically stable as compared with the hotspot mutants. In fact, the Ea of the two temperature-sensitive mutants, P223L and V274F, were essentially similar to the Ea of wild type, suggesting similar thermodynamic stabilities. The same data (Table 1) plotted with wild-type set as a reference point clearly show the magnitude of energy differences for various core domain mutants (Fig. 2B). The ΔEa between the wild type and R175H and R273H have more difference in minimized energy to the wild type and with temperature-sensitive mutants.

Na = number of atoms in the simulation.
The stability of wild-type p53 core domain is 6.0 kcal (1 kcal = 4.18 kJ)/mol at 25°C and 9.8 kcal/mol at 10°C based on denaturation curves measured by differential scanning calorimetry/spectroscopy (Bullock et al., 1997), suggesting that the p53 core domain is of modest thermodynamic stability. Based on the above method, the calculated equilibrium denaturation of p53 core domain at 10°C was −2.97 kcal·mol−1·M−1 (Bullock et al., 1997). The calculated equilibrium denaturation at 10°C of p53 mutants R175H, C242S, R248Q, R249S, and R273H was −2.59, −2.68, −2.91, −3.09, and −3.11 kcal·mol−1·M−1, respectively (Bullock et al., 1997). Except R249S and R273H, the equilibrium denaturation at 10°C of p53 mutants was lower than that of the wild type, possibly because these mutants appear to be part of the DNA binding domain. In contrast, the simulated normalized (per atom) calculated energy difference in our study at least for R273H is less than the respective wild-type p53 core domain (Table 1 and Fig. 2). Functionally, R273H is also a gain-of-function mutant found in the majority of cancers.
As compared to the temperature-sensitive mutants, the effect of mutating an arginine is noticeable; changing an arginine to a histidine in two different positions shows the similarity of impact on minimized energy. Therefore, to understand whether the energy differences were specifically caused by mutating arginine to histidine, we simulated the minimum energy calculations using another arginine mutation, R248Q. R248Q is also a common mutation in many cancer types. Table 2 shows the minimized energy (Ea) of R248Q. The energy difference between R248Q and wild type in the context of R273H and R175H, shown in Figure 2B, suggests that the arginine residue places wild-type p53 in lower energy with respect to the mutants. As such, it is not surprising that majority of mutations involve the arginine residue that alters the minimum energy of p53. Thus, irrespective of whether arginine mutations are structural (R175H) or functional (R273H) (Joerger and Fersht, 2008), targeting an arginine residue appears to play a very significant role in the thermodynamic stability of the p53 core domain.
We hypothesized that replacing arginine, the third heaviest residue in terms of molar mass, by a lighter residue may decrease thermodynamic stability. To study the effect of mutating a residue on the overall energy, we focused on linking the specific mutation to the overall bonded terms in energy. As stated in the Methods section, we used μ, a dimensionless quantity, to establish a correlation between the mutated and the wild-type protein. Table 3 summarizes μ results for p53 core domain mutants that are also shown in Figure 3.
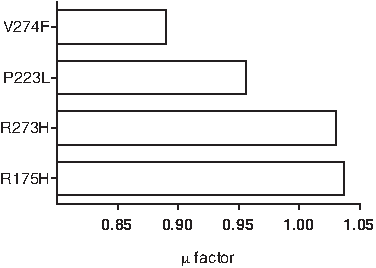
Approximation (μ) factor in correlation between residue molar masses and potential energies for mutated proteins.
A comparison between approximation in Figures 2 and 3 indicates that two different methods place mutated proteins in a similar context as compared to the wild type. Because μ was a calculated approximation of mutants and its two immediate neighbors with an approximation similar to that achieved by simulations in CHARMM, we used a similar approximation for additional core domain p53 mutants found in various cancers (Table 4).
The minimum amount of prediction, G245D mutated protein is underlined. Amino acids in bold refer to the wild type (column 3) and the corresponding mutated amino acid (column 4).
The results indicate that changes in potential energy depend on the molar mass of not only the target residue, but also at least two other residues near the target residue. If two different mutations label the same residue position (e.g., R248Q and R248W), replacement by the lighter residue (Q) in molar mass sets protein at the higher potential energy. Moreover, when the target and replacement residues are the same (R175H and R273H), in the different positions, then changing the potential energy will be determined by the molar mass of near-neighbor residues. Mutations may present similar results (R248W and R282W) in our approximation if two neighbor residues are very similar or the same in molar masses. Every mutation with μ > 1 in our results referred to a higher frequency in human cancer in p53. The rate of mutation in p53 in cancer references compares to the μ factor (Table 5). Our approximation, therefore, matches to p53 mutations in cancer (Freed-Pastor and Prives, 2012) adequately.
The minimum percentage of mutation in biology references matches to our prediction (underlined mutated protein).
3.1. Solvation
All the energy calculations described above are for minimized protein structure in the vacuum as an approximation to reduce noise. Because the proteins rarely exist in a dehydrated/vacuum space, we therefore calculated the energies of the minimized structure of wild-type and four mutated p53 core domains solvated in a box of water as large as the dimensions of each protein individually (block of water) (Fig. 4). The calculated energy in the vacuum and solvation for the wild-type and mutant proteins was compared (Table 6). The results suggested that there is no significant change in the calculated energy in the vacuum as compared to the block of water.
Wild-type protein
E(1) = energy of protein in the vacuum. E(2) = energy of protein in the box of water.
4. Conclusions
The molecular dynamic simulation of mutant forms of p53 can be predictive and correlative of the rate of mutation in human cancer. Consequently, further investigations may be required before these findings can be generalized and applicable to diverse p53 mutants. Nonetheless, the R175H and R273H mutations are highly relevant to human cancers as well as many other hotspot mutations studied herein. A typical search of the IARC database (Oliver et al., 2002) establishes the prevalence of R175H and R273H as a somatic mutation in numerous different tumors and as a germline mutation in families with Li-Fraumeni syndromes.
The less frequently occurring p53 mutations (P223L and V274F used in this study) have been shown to act essentially as wild type at lower temperatures, suggesting intrinsic flexibility. It is therefore not surprising that the calculated energies of these two mutants are similar to the wild-type p53. Thus, taken together, our studies reveal that p53's intrinsic flexibility can yield promising therapeutic avenues for the restoration of mutant p53 biological processes. Thus, manipulation of missense mutations found in p53 via simulation can be predictive of the rate of frequency of p53 mutations and provide an insight to what therapeutic avenues could be most suitable for p53 restoration.
Footnotes
Acknowledgments
This work was supported in part by the National Science Foundation (Grants DMR-0934142, XSEDE TG-DMR120078, and HRD-11137751, to L.R. and X.-Q.W.).
Author Disclosure Statement
The authors declare that no competing financial interests exist.