
Abstract
Abstract
At the core of Illumina's high-throughput DNA sequencing platforms lies a biophysical surface process that results in a random geometry of clusters of homogeneous short DNA fragments typically hundreds of base pairs long—bridge amplification. The statistical properties of this random process and the lengths of the fragments are critical as they affect the information that can be subsequently extracted, that is, density of successfully inferred DNA fragment reads. The ensembles of overlapping DNA fragment reads are then used to computationally reconstruct the much longer target genome sequence. The success of the reconstruction in turn depends on having a sufficiently large ensemble of DNA fragments that are sufficiently long. In this article using stochastic geometry, we model and optimize the end-to-end flow cell synthesis and target genome sequencing process, linking and partially controlling the statistics of the physical processes to the success of the final computational step. Based on a rough calibration of our model, we provide, for the first time, a mathematical framework capturing the salient features of the sequencing platform that serves as a basis for optimizing cost, performance, and/or sensitivity analysis to various parameters.
1. Introduction
R
1.1. High-level description of the problem
A target DNA strand to be sequenced can be viewed as a possibly long, for example, 109, sequence of L letters. In shotgun sequencing, a large number of copies of the target are first randomly cut into fragments; the fragments are then sequenced, each providing a read of length l, where l is much smaller than L (Venter et al., 1996, 1998). The approach involves two key steps. In Step 1, one reads as many fragments as possible—as we elaborate later in this section—that can be parallelized and therefore performed very efficiently. In Step 2, one attempts to reconstruct the target sequence by leveraging the library of overlapping reads obtained in Step 1—this step is referred to as assembly. For clarity, let us consider the two steps independently, although one should keep in mind that they are intimately linked and will, in the sequel, be jointly optimized.
To functionalize the surface of a DNA chip (referred to as a flow cell), DNA fragments are first scattered across its surface whereupon they attach at random locations (Bentley et al., 2008). A single fragment is insufficient to generate a signal that is detectable; to remedy this, each of the initially positioned fragments, which we refer to as germs, is replicated in parallel a number of times through the process called bridge amplification. The resulting ensembles of fragments, each comprising hundreds of identical copies of a germ, enable signal amplification and accurate DNA sequence detection. The germ replication can be viewed as a spatial branching process happening on the surface of the flow cell. The result of each such process, in the simplest case, is roughly a disc of the fragment's copies centered at the location where the original strand (germ) happened to attach to the flow cell. The radius of the disc can be controlled by the number of steps of the bridge amplification process. As we shall see, due to possible interaction among such growth patterns, the resulting shapes might be more complex than discs, so in the sequel, we will more generally refer to them as clusters.
As mentioned above, each fragment is replicated to generate a cluster of identical molecules, which enables signal amplification and thus facilitates sequence detection, that is, the underlying letter reading mechanism. Reads are obtained in parallel, that is, all clusters are read simultaneously one letter at a time. This is accomplished by relying on reversible terminator chemistry (Bentley et al., 2008) where the first unread letter of each fragment is identified by detecting the color of the fluorescent label attached to the nucleotide bound to it. 1 Since clusters contain multiple copies of the fragments, with proper illumination, each cluster will light up the color indicative of the latest letter/base being examined. This process, referred to as sequencing-by-synthesis, is applied sequentially for say l steps and thus, in principle, one can determine the first l letters of all fragments on the flow cell—this is the aim of Step 1.
There are two caveats, however. First, the successive reading of nucleotides can fail for some fragments in each cluster. Specifically, on a given step, say k, the chemical processes associated with reading the kth nucleotide base may fail or jump ahead to the (k + 1)st one. Thus, at each step, only a fraction of the fragments in a cluster have their kth nucleotide properly marked with the correct fluorescent marker. As this proceeds, an increasing number of markers get out of phase, that is, the disc/cluster will eventually appear to have a mix of colors, making correct detection of the fragment's next letter increasingly difficult. To deal with this phenomenon, typically referred to as phasing, a number of base calling methods have been proposed in recent years (Erlich et al., 2008; Kao et al., 2009; Kircher et al., 2009; Kao and Song, 2011; Das and Vikalo, 2012, 2013). Indeed, amplifying the signal is the reason for synthesizing the cluster of duplicates of each fragment in the first place, that is, clusters are meant to enable in-phase addition of light emanating from a number of identical fluorescent tags.
The second caveat is that randomly placed germs may be grown into clusters that overlap, which will also impair the reading process. Larger clusters will tend to experience more overlaps. In essence, this is a random disc packing problem: if two germs happen to attach to the flow cell at distance r from each other, any cluster growth that leads to discs of radius larger than r/2 leads to such an overlap and hence to an impaired reading of the letters of the corresponding two strands.
We are now in a position to articulate the main trade-offs that drive the efficiency of shotgun sequencing, which assembles the target using short reads from a flow cell. There are three main parameters at play: the density λ of fragments initially placed on the flow cell, the length l of the fragment reads, and the disc radius r associated with the bridge amplification process:
• λ large looks desirable (because more fragment reads will facilitate Step 2), but could be problematic for any fixed r because of possible disc overlaps; • l large is desirable (because it will facilitate Step 2), but could be problematic because of the deterioration in read quality as base pair reads get out of phase; • r large is necessary (to amplify signals and facilitate detection), but this precludes the desire for a large λ (because of disc overlaps).
We pose the following question: What is the optimal set of parameters to maximize the yield and/or possibly minimize the cost in the presence of all these trade-offs?
There are many ways to frame the problem of optimizing yield in such systems. In this article, we will first consider optimizing fragment yield, which we define as the density of length l fragments successfully read per unit area of the flow cell. Then, we consider metrics that are more directly tied to the final objective of sequencing length L target DNA sequence. To that end, let us consider Step 2, the reassembly process.
Following a simple stylized model of the process, a condition for successful reassembly is that the collection of successfully read fragments covers the target DNA—that is, if one denotes by
A critical trade-off associated with Step 2 now emerges. It is between the number of correctly read fragments and their length. A large collection of fragments is helpful, but if they are too short, that is, l is small, it is difficult to obtain a covering of the entire DNA target sequence 2 .
This brings us to the main problem addressed (and solved) in this article. The goal is optimize the key parameters associated with the sequencing process we described earlier (namely the density λ of fragments placed on the flow cell, the duration of the growth process, and the length of the fragment reads l) so as to ensure a prespecified probability, say δ, for example, 99%, of obtaining a covering of the target DNA sequence at minimal operational cost. Operational costs can be viewed as being in two main categories: (1) those associated with raw materials, for example, DNA copies, reagents, and flow cells; and (2) those associated with time, for example, the time spent on the sequencing machine or the execution time of the signal processing algorithms. In this article, we shall for simplicity adopt the flow cell area as the cost. Some reagent costs and some flow cell processing time costs might be proportional 3 to the flow cell area. Given an initial rough calibration of the model based on available data, we are able to show how to optimize the steps/parameters of the sequencing process. We stress, however, that the article provides a general mathematical framework, which can accommodate other costs and objectives, for instance, costs that are proportional to the total number of strands (this might be the case for certain processing times) or to the number of DNA copies (raw material) rather than to the area. We will not discuss them here for the sake of brevity.
1.2. Related work
To our knowledge, this is the first work attempting to model and analyze the cluster growth process with a view on optimizing DNA sequencing cost/yield. The detailed simulations of the surface physics associated with the bridge amplification process (Mercier et al., 2003; Mercier and Slater, 2005) support that the disc/cluster processes we introduced earlier and will use are well suited. Work optimizing this process has taken place in industry where empirical evidence and simple rules of thumb have been used. There is, however, a substantial body of work toward developing mathematical tools for analyzing random spatial processes [see, e.g., (Stoyan et al., 1995) and some of our work (Baccelli and Blaszczyszyn, 2009)]. Indeed, this branch of mathematics is now ubiquitous with applications in material science, cosmology, life sciences, and information theory, to name a few. Further developments of the mathematical foundations have recently been carried out by us in Baccelli and Blaszczyszyn (2009) and proven to be invaluable, for example, to understand fundamental characteristics of large wireless systems and optimizing their performance. Such stochastic geometry models have been embraced by academia and industry, providing insight into current and future technological developments. Indeed, the aim of this article is to show that this may also play a role in the DNA sequencing setting.
As mentioned earlier in this section, sequence assembly may be performed with or without referring to a previously determined sequence (genome, transcriptome). De novo genome shotgun assembly is a computationally challenging task due to the presence of perfect repeat regions in the target and by limited lengths and accuracy of the reads (Miller et al., 2010; Bresler et al., 2013; Motahari et al., 2013). In the reference-guided assembly setting, the reads are first aligned (i.e., mapped) to the reference, easing some of the difficulties faced by the de novo assembly (Delcher et al., 2002). However, the reference often contains errors and gaps, creating a different set of challenges and problems. In fact, if the sample is highly divergent from the reference or if the reference is missing large regions, it may even be preferable to use de novo assembly (Iqbal et al., 2012). While the development of methods for sequence assembly received significant attention, ultimate limits of their performance have been less explored. The pioneering work of Lander and Waterman (1988) provided the first simple mathematical model for the reassembly process. This has been followed with various refinements (Bresler et al., 2013; Motahari et al., 2013), but to our knowledge, none has provided a mathematical framework to compute the flow cell parameters needed to achieve a given likelihood of full target coverage. Moreover, no previous work has linked this end objective to the optimization of the end-to-end process as we will do in this article.
In Wendl and Wilson (2008) and related articles, the authors derive closed-form expressions for the first and second-order statistics of the sequencing depth, which pertains to the number of reads covering bases along the target. Specifically, the expected sequencing depth and its variance are expressed in terms of the parameters of the sequencing task (target and read length, number of reads) while taking into account practical issues, including edge effects and the paired-end nature of the reads. Interestingly the authors find that due to these effects, for small to moderately long targets, shorter reads may provide higher coverage per unit sequence depth than longer ones. This notion of sequencing depth can be seen as a coverage problem in the sense of Hall (1988) and Stoyan et al. (1995). However, this coverage problem is one dimensional (linked to what we call the reassembly problem below) and is quite different from the two-dimensional coverage processes taking place on the flow cell, which are analyzed in the present article, for example, the Boolean model (see again Hall, 1988; Stoyan et al., 1995) discussed below.
The article is organized as follows: we first describe the stochastic geometric models for the distribution of clusters on the flow cell resulting from the bridge amplification process. We then analyze the impact of the geometry of clusters on the achievable yield of fragment reads and discuss its optimization. The last section proposes a simplified model for the reassembly problem, which is based on a queuing model. This allows us to consider the end-to-end cost optimization of the sequencing process to meet a desired likelihood of coverage for the target DNA sequence.
2. Stochastic Geometry for Shotgun DNA Sequencing
In this section, we introduce basic geometric models for the cluster processes associated with the DNA fragments resulting from the bridge amplification procedure on the surface of the flow cell. These are the singleton cluster, shot-noise, and Voronoi models, respectively. These processes will be tied to the salient features of fragment-reading mechanisms.
In the singleton cluster process model, all clusters that intersect (or touch) another cluster are discarded. The retained clusters are roughly modeled as discs of radius r consisting of duplicates of the same DNA fragment. In the shot-noise process model, an attempt is made to read each cluster. Isolated clusters are as in the singleton cluster case. A cluster, which is in contact with one or more clusters, is still analyzed as a disc of radius r; however, depending on the number and shape of the other clusters in contact, part of the light signal stemming from that disc creates an interference, which is treated as noise. If signal dominates interference/noise, one can still read this cluster. Finally, the Voronoi case studies are the (hypothetical and somewhat futuristic) scenarios where one computes optimal masks that allow one to mask all clusters in contact with the tagged cluster and hence to cancel interference.
Some of these models will be used for the reassembly optimization alluded to above. For each case, we describe the mathematical approach used to evaluate its performance. This will also be used to give some yield optimizations of independent interest.
2.1. Random seed model and growth model
We consider the locations of the initial DNA fragments (the centers of the clusters or the seeds) to be a homogeneous Poisson point process N on
If it does come into contact with another cluster, we assume the growth in that direction stops, but growth continues in all other directions. As r approaches infinity, the configuration of clusters becomes the Voronoi diagram associated with the point process N (Stoyan et al., 1995). For r finite, the shapes are known as the Johnson–Mehl growth model (Stoyan et al., 1995).
2.2. Read reliability model
As already explained, phasing problems occur because nucleotides occasionally fail to incorporate in particular duplicates or anneal to the base pair right next in line. These duplicates are then out of sync with the rest of the duplicates and give off a different color signal. So, even though amplifying the fragments gives a much stronger signal, there is noise due to these out-of-phase duplicates limiting the accuracy of the reads.
To model this in a single cluster, let Xl denote the number of copies of the original DNA fragment that remain in phase after l steps of the process. Let p be the probability that a DNA fragment gets out of phase at one step.
The random variable Xl has a binomial distribution, where the number of trials is the number of DNA fragment copies in a cluster and p(l):=(1 − p) l is the probability that a fragment remains in phase after l steps, that is, the probability of success.
At first glance, it makes sense to require that
2.3. The singleton cluster model
For a cluster with unimpeded growth over time r, the number of DNA fragments in a cluster is aπr2, so
When the number of trials is large, the binomial distribution can be approximated by the normal distribution. In the singleton case
A cluster centered at
The overall fragment yield of singletons is the intensity of the isolated clusters times the probability a cluster will enjoy a correct read:
2.4. The shot-noise model
Using only isolated clusters is clearly suboptimal. We consider here the situation where all clusters are used. In this case, for each cluster, the amount of interference from contact with other clusters during the growth/duplication process has to be taken into account. According to our growth assumptions, the area of the typical cluster (assumed with a seed located at 0) is
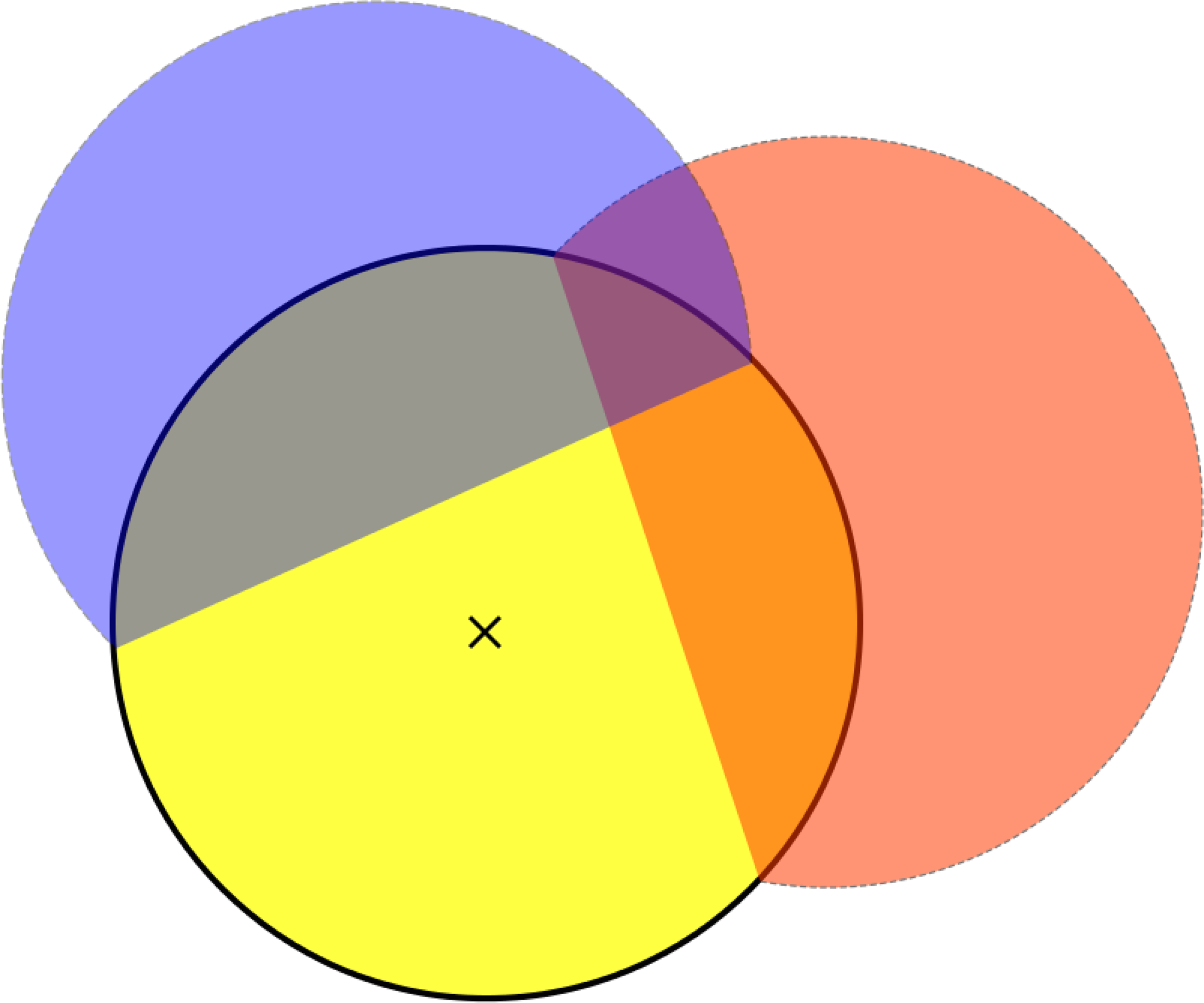
Cluster interference.
This interference upper bound can be described in terms of a shot-noise field
The Laplace functional of the interference is as follows:
Overlap with the typical cluster
where
The random variable
and the fragment yield is
where f is the law of I, which is known through its Laplace transform (Stoyan et al., 1995). The computation of this yield, which is based on Fourier techniques, is discussed in the Appendix of O'Reilly et al. (2015).
2.5. The Voronoi model
In this subsection, we consider an optimal scenario for collecting signal reads using our assumptions about cluster growth. Clusters are allowed to grow until they have formed a Voronoi tessellation. Then, optimal sized masks, having the shape of each Voronoi cell, are used to read the signal. In this scenario, no interference from neighboring clusters is present, and the only variable to optimize is the intensity λ of the underlying point process.
The closed form of the distribution for the area of Voronoi cells is unknown, but it can be approximated by the generalized Gamma density:
This is the approximate distribution for the normalized cell size λA, where A denotes the area. For λ = 1, good choices for the area are γ = 1.08, ν = 3.31, and χ = 3.03 (Stoyan et al., 1995).
For a general intensity λ, the area distribution is given by
for x ≥ 0, where γ = 1.08, ν = 3.31, and χ = 3.03. The expected fragment yield for the Voronoi case is then
3. Fragment and Letter Yield
In this section, we first study the fragment yield, namely the mean number of correctly read fragments per unit space of the flow cell. We then study the letter yield, which is based on the number of letters correctly read.
3.1. Numerical results on fragment yield
We use the mathematical expressions obtained above to optimize the yield in all three models for l fixed. For the singleton cluster and the shot-noise model, we optimize over the radius r and the intensity λ. For the Voronoi model, the optimization is over λ. Tables 1 and 2 show optimal parameters and fragment yields for l = 200 and l = 150.
For l = 200, a 45.7% increase in optimal fragment yield can be obtained when considering all clusters and not just the singleton ones. This increase in yield comes with a slightly larger radius and higher intensity than the optimal parameters for singleton clusters. This corresponds to increasing the amount of time for replication and increasing the number of initial DNA fragments spread over the flow cell. This will mean more clusters overlap with their neighbors, but many more correct reads can still be made for clusters that only run into their neighbors late in the growth stage.
For each case, decreasing l to 150 from 200 resulted in a smaller optimal radius and a larger optimal intensity. With a smaller l, a fragment is more likely to remain in-phase, making it easier for in-phase fragments to comprise half the cluster plus the fixed margin ε. This allows clusters to be smaller and more densely packed to obtain a higher yield.
The percent increases in the fragment yield obtained when switching to l = 150 are for Singletons: 221.59%, for Interference: 278.87%, and for Voronoi: 202.96%.
3.2. Numerical results on letter yield
In view of the differences between l = 200 and l = 150, it makes sense to consider the optimal letter yield, namely lλy(l, r, λ), the mean number of letters correctly deciphered per unit space. So, above, optimization takes place w.r.t. l as well.
We see in Table 3 that the optimizing value of l is actually somewhere around 100, which is shorter than the read length provided by, for example, Illumina's HiSeq sequencing platforms.
The percent increases in the fragment yield obtained when switching from l = 150 to the optimal l are for Singletons: 31.07%, for Interference: 49.14%, and for Voronoi: 18.89%. In this optimized setting, the letter yield of the shot-noise case is close to twice that of the singleton cluster case.
4. Reassembly Model and Optimization
This section is focused on the optimization of the probability of reassembly of the original DNA sequence.
4.1. Reassembly model
The reassembly question can be formulated in terms of n, the number of fragments in the genomic library (fragments correctly deciphered); L, the DNA sequence length in base pairs (for the human genome, L = 3 billion); and l, the length of the fragments in the same unit. We see L as a segment on the real line and we assume that the fragment starting points form a Poisson point process on the real line with parameter
As already explained, we are interested in the probability of complete reassembly. We will first reduce this to the probability that all letters are covered.
4.2. Analytic expression for the reassembly probability
Within the Poisson setting described above, this can be reduced to a queuing theory question: consider an M/D/∞ queue, namely a queue with Poisson arrivals, an infinite number of servers, and a constant service time of length l. The probability of reassembly is then just the probability that the busy period in such a queue exceeds L. The distribution of the busy period can be determined through its Laplace transform.
Let
The busy period of the queue is
where
Now, if
Since
We want to compute
4.3. Reassembly optimization
In our optimization, we ask the following question: For a given genome length, what is the smallest area the sequencing flow cell needs to have to get a high probability that the entire genome can be reassembled?
To answer this, let
For each
Finally, we optimize over
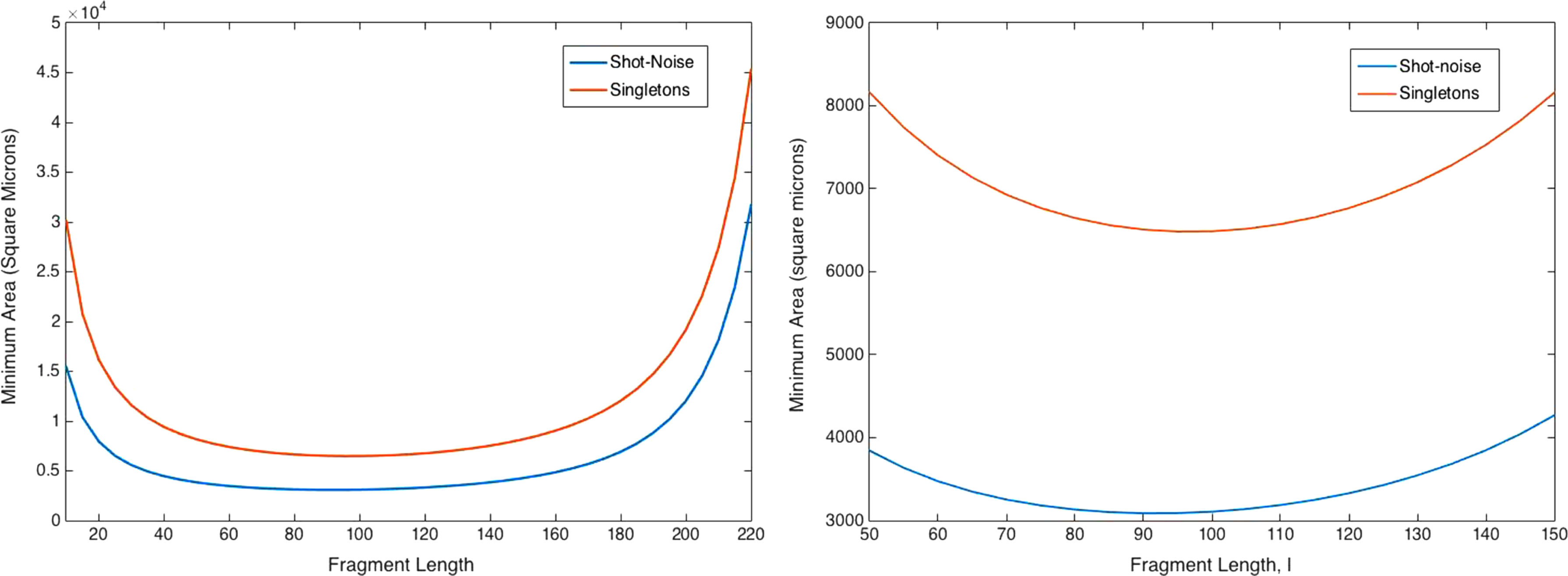
Minimum area needed to achieve a probability of reassembly of 0.99 versus the length of the fragments for a genome of length 100,000.
5. Conclusion
This article establishes a connection, which is new to the best of our knowledge, between stochastic geometry and queuing theory on one side and fast DNA sequencing on the other side. This connection allowed us to propose a simple model, which captures the key steps of the fast sequencing process: segmentation of multiple copies of the DNA into random fragments, replication of the randomly placed fragments on the flow cell, spatial interactions between the resulting fragment clusters, read of fragments through their cluster amplification, taking the possibility of read errors and interference between clusters into account, and finally assembly of the successfully read fragments. This model is analytically tractable and allowed us to quantify and optimize various notions of yield, including the yield of the end-to-end sequencing process, in function of the parameters. This basic model seems generic and flexible enough for us to envision a series of increasingly realistic and yet tractable variants for each step of the process and eventually a comprehensive quantitative theory for this class of sequencing problems.
Footnotes
Acknowledgments
The work of the first two authors was supported by a grant of the Simons Foundation (#197982 to UT Austin). The work of the first author was supported by the National Science Foundation Graduate Research Fellowship (Grant DGE-1110007).
Author Disclosure Statement
No competing financial interests exist.