
Abstract
Abstract
Protein–ligand docking can be formulated as a search algorithm associated with an accurate scoring function. However, most current search algorithms cannot show good performance in docking problems, especially for highly flexible docking. To overcome this drawback, this article presents a novel and robust optimization algorithm (EDGA) based on the Lamarckian genetic algorithm (LGA) for solving flexible protein–ligand docking problems. This method applies a population evolution direction-guided model of genetics, in which search direction evolves to the optimum solution. The method is more efficient to find the lowest energy of protein–ligand docking. We consider four search methods—a tradition genetic algorithm, LGA, SODOCK, and EDGA—and compare their performance in docking of six protein–ligand docking problems. The results show that EDGA is the most stable, reliable, and successful.
1. Introduction
P
The scoring function is a free energy of binding interaction between protein and ligands. Scoring function can help a docking to efficiently explore the binding space of a ligand. It is also responsible for evaluating the binding affinity once the correct binding pose is identified (Bharatham et al., 2014; Li et al., 2015).
The search algorithm for solving the docking problem of flexible docking aims to identify the docked conformation with the lowest energy. Some metaheuristics (Blum et al., 2011; López-Camacho et al., 2015) have been successfully applied in the docking problem, and many researchers have tried improving the optimization algorithm of protein–ligand docking. For instance, simulated annealing (SA) (Goodsell and Olson, 1990) is a generic probabilistic metaheuristic for the global optimization problem of locating a good approximation to the global optimum of a given function in a large search space. Genetic algorithm (GA) (Jones et al., 1997; Morris et al., 1998; Thomsen, 2003; Cao and Li, 2004) is a method to search the optimal solution by simulating the natural evolution process. Lamarckian genetic algorithm (LGA) (Fuhrmann et al., 2010) is a hybrid of GA and the local search, and it is more successful than SA and GA for the docking problem. SODOCK (Chen et al., 2007; Jason et al., 2008) based on particle swarm optimization (PSO) integrates with a local search, and it is designed for flexible docking.
There are two criteria used to verify the performance of different optimization algorithms: fitness accuracy (energy based) and pose accuracy (root-mean-square deviation [RMSD] based) (Guo et al., 2014). For fitness accuracy, the lower binding energy is the greater binding activity that can also provide better drug efficiency. RMSD is used to determine if two docked conformations are similar enough to be included in the same cluster. A docked conformation with a smaller RMSD is considered as a more accurate solution to the docking problem. Based on these two standards, the existing algorithms are proven to have obvious shortcomings. Therefore, an efficient optimization algorithm that can find lower docking energy and RMSD is desirable.
The novel algorithm is improved on the basis of LGA. LGA is proven to be efficient, but its search direction is blind random. Because function evaluation of solving the lowest energy is the most computational-intensive process in the search algorithms, such randomness is clearly a waste of computational resources. Furthermore, LGA has no effective use of feedback information, and so its search speed is slow and it cannot obtain a more accurate solution. To improve LGA, a modified LGA that has evolutionary direction is presented in this article to solve the aforementioned docking problem.
The implementation of EDGA adopts the environment and scoring function of AutoDock 4.2.6 (Morris et al., 1996, 2009; Kitchen et al., 2004). AutoDock is the most widely used automated docking program. To evaluate the method, we perform six protein–ligand docking problems from the Brookhaven Protein Data Bank (PDB) (Berman et al., 2000, 2002). In this article, we compared the performance SA, GA, SODOCK, and EDGA. Computer simulation results reveal that EDGA is superior to the other methods in terms of convergence performance, robustness, and obtained energy, especially for highly flexible ligands. Simulation results also reveal that EDGA can yield more accurate results than the other methods in terms of RMSD.
2. Scoring Function
AutoDock 4.2.6 uses a semiempirical free-energy force field to evaluate a docked conformation. The force field includes six pair-wise evaluations (V) and an estimate of the conformational entropy lost upon binding (Δ Sconf):
where L represents the ligand and P represents the protein. Each of the pair-wise energetic terms is expressed as the sum of dispersion/repulsion in which the parameters are based on the Amber force field, hydrogen bonding, electrostatics, and desolvation.
3. Methods
3.1. Hybrid search of EDGA
In this article, we propose a novel algorithm to high protein–ligand docking. The new algorithm based on LGA integrates with a guided evolutionary direction mechanism so that solutions are in a better direction. The algorithm will be abbreviated as EDGA, which stands for a Population Evolution Direction-Guided Genetic Algorithm. EDGA is considered as a genetic algorithm module (GAM) cooperating with the standard GA operators such as population initialization, crossover operator, and selection operator, while evolution direction module (EDM) consists of the mutation, the guided evolutionary direction, and applying the local search. Figure 1 shows the block diagram of EDGA.
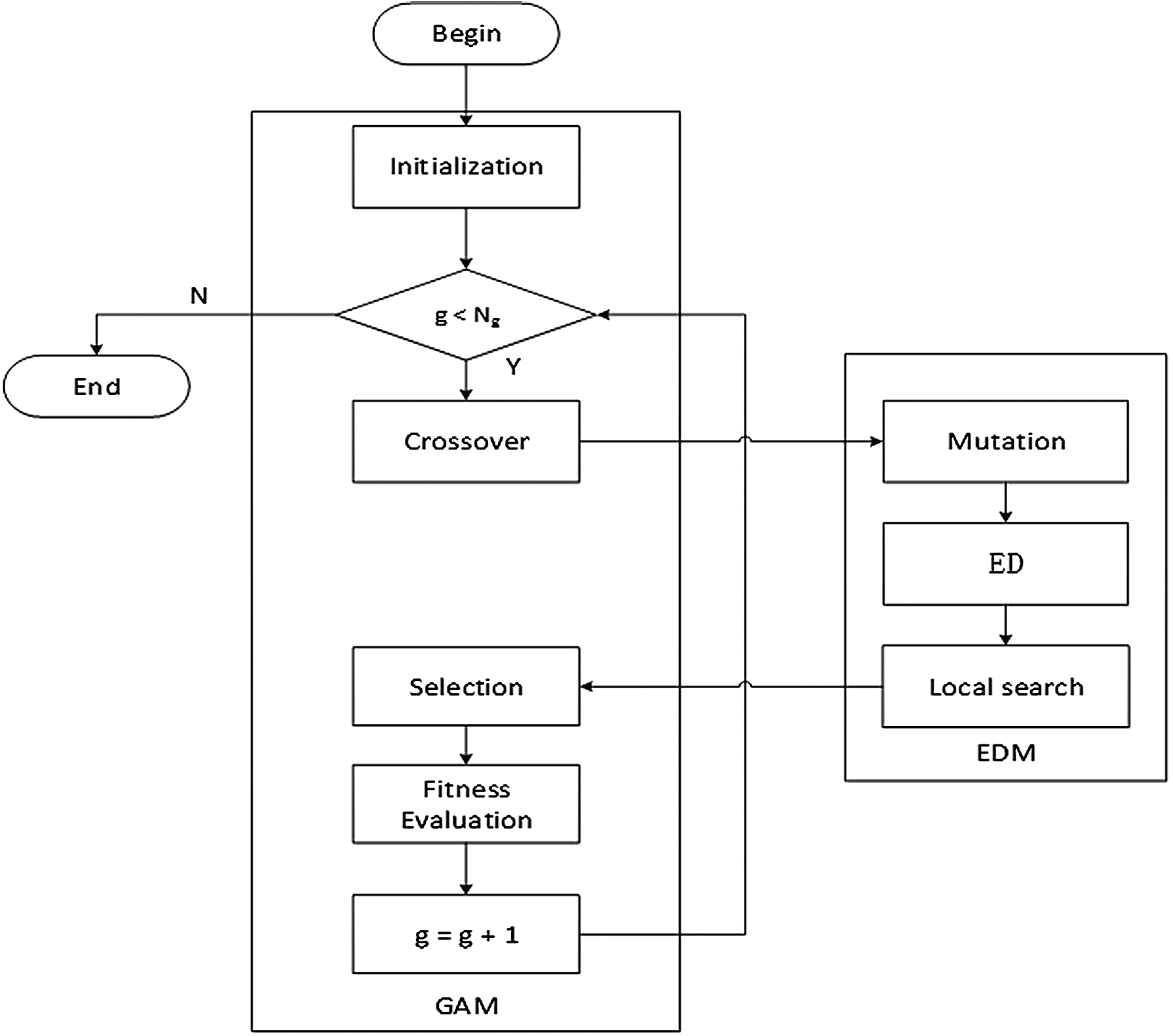
Block diagram of EDGA.
EDGA starts with initializing the population. Afterward, the offspring population is generated by genetic operators such as crossover and mutation. The new population is then selected from the parent population and the offspring population. The reproduction process and the selection process are repeated until the number of iterations exceeds a predetermined value.
3.2. Evolutionary direction
In the original algorithm, the mutation is random and the search direction is blind random. In the early stage, the randomness plays a very good guide effect for the global search. The direction of the optimal solution is not known. With the development of the algorithm, the search experience is accumulated, the direction is gradually clear, the search space begins to converge, and the difference between each solution is getting smaller, which means inevitably that the abandoned solution is the current best—even the best. When the algorithm search space reduces to a very small range, if you continue to take the random pattern of the original algorithm, then it is possible to lead away from the search target. At this time, the search strategy needs to be adjusted. Therefore, a new mutation method is introduced.
In the method, an equilibrium factor β is introduced. When the random factor φ < β, the random mutation is used; otherwise, the “mixed mutation” is used. This new mutation combines the information provided by the optimal solution and the suboptimal solution of the history.
In the improved algorithm, the mutation operator will be generated according to the following formula:
where xij is the solution vector of the current value, φ is a random number between 0 and 1, β is a particular adjustable parameter (the range is 0 to 1), xoptimum represents the solution vector of the historic optimal solution, and x sub represents the solution vector of the historic suboptimal solution.
In the formula, the purpose of introducing the current solution and the second best solution is to keep the search direction to the optimal solution as far as possible. In principle, as shown in Figure 2, A is the current optimal solution, B is the historic suboptimal solution, and C is the historic optimal solution. Thus, φ (C − A) + φ (C − B) = φ AD + AC = AE, where φ is 0 to 1. As a result, the search direction will be closer to the AE direction because the historic optimum solution is in the vicinity of the direction, and so the possibility of finding the global optimal solution becomes larger. On the other hand, if the current optimal solution is the historic optimal solution, that is, when C and A coincide in the schematic diagram, the AE direction is coincident with the BC direction. The actual moving direction of the search is changed into AD. In this case, the optimal solution of the group is abandoned, but it can also guarantee the correctness of the direction.
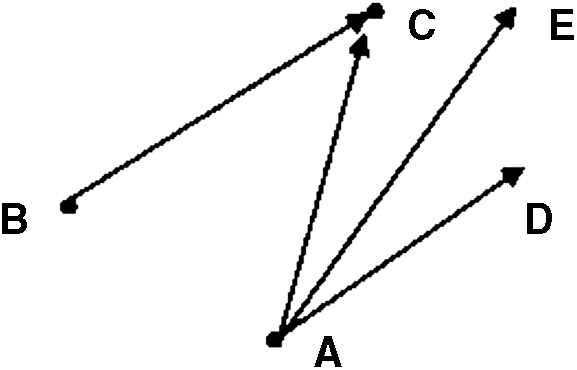
Schematic diagram of GE.
4. Experiments and Discussion
Six protein–ligand complexes (Hu et al., 2004) were chosen from the Brookhaven PDB (Berman et al., 2000, 2002) to compare the performance of the docking techniques. The six docking problems are summarized in the following:
(1) β-Trypsin/Benzamidine (3ptb)
Benzamidine is a reversible competitive inhibitor of trypsin, trypsin-like enzymes, and serine proteases. The recognition of benzamidine by β-trypsin is mainly because of the polar amidine moiety and the hydrophobic benzyl ring.
(2) McPC-603/Phosphocholine (2mcp)
Phosphocholine is an intermediate in the synthesis of phosphatidylcholine in tissues. The recognition of Phosphocholine by FabMcPC-603 is mainly because of the influence of ArgH52.
(3) Streptavidin/Biotin (1stp)
Biotin, also known as vitamin H or coenzyme R, is a water-soluble B vitamin. Streptavidin/biotin is one of the most tightly binding noncovalent complexes.
4) HIV-1 Protease/XK263 (1hvr)
The cyclic urea HIV-protease inhibitor, XK-263, has 10 rotatable bonds, excluding the cyclic urea's flexibility.
5) Influenza Hemagglutinin/Sialic Acid (4hmg)
The recognition of sialic acid by influenza hemagglutinin is chiefly because of hydrogen bonding.
6) Dihydrofolate Reductase/Methotrexate (4dgr)
Methotrexate is an antimetabolite that attacks proliferating tissue and selectively induces remissions in certain acute leukemias.
We compared the performance of GA, LGA, SODOCK, and EDGA. The semiempirical free-energy force field presented above was used for energy evaluation in all cases. The main goal was to find the lowest energy in each docking problem. We also compared the root-mean-square positional deviation (rmsd) between the lowest energy docked structures. The rmsd tolerance was used to determine if two docked conformations were similar enough.
It is important to ensure that different search methods are treated equally. Therefore, in the three GAs, the population was 50, the number of generations was 27,000, and the energy evaluations was 1.5 × 106 in a docking. Therefore, the dockings were terminated by reaching the maximum number of generations. In SODOCK, the number of particle was 50, the number of immediate neighbors was 5, and the maximal number of function evaluation was 1.5 × 106. Five times were tested and each time consisted of 10 runs, and so there were 50 results.
The results of GA, LGA, EDGA, and SODOCK docking experiments are summarized in Tables 1–4, respectively. Through these tables, we concluded that EDGA found the lowest energy in five of the six protein–ligand docking problems. The number of the lowest energy corresponding to the lowest rmsd was 1, 1, 2, and 2 for GA, LGA, EDGA, and SODOCK, respectively. Thus, EDGA performed best in finding the lowest energy docked structure and the lowest rmsd. Furthermore, the number of the lowest mean energy was 0, 0, 4, and 2, using GA, LGA, EDGA, and SODOCK, respectively. However, the number of the lowest mean rmsd found by each of the four search methods was 1, 2, 2, and 1. In conclusion, considering their average performance, the best search method was EDGA.
PDB, Protein Data Bank; rmsd, root-mean-square positional deviation.
Figures 3–8 are convergence diagrams, and these figures show that EDGA also had the best convergence performance among all methods. Figures 9–14 are box plots, and the median of EDGA was the lowest in five of the six docking problems and the data of EDGA were the most concentrated. Tables 5–10 are hypothesis tests. Through these tables, it can be seen that EDGA performed significantly better than LGA, GA, and SODOCK.
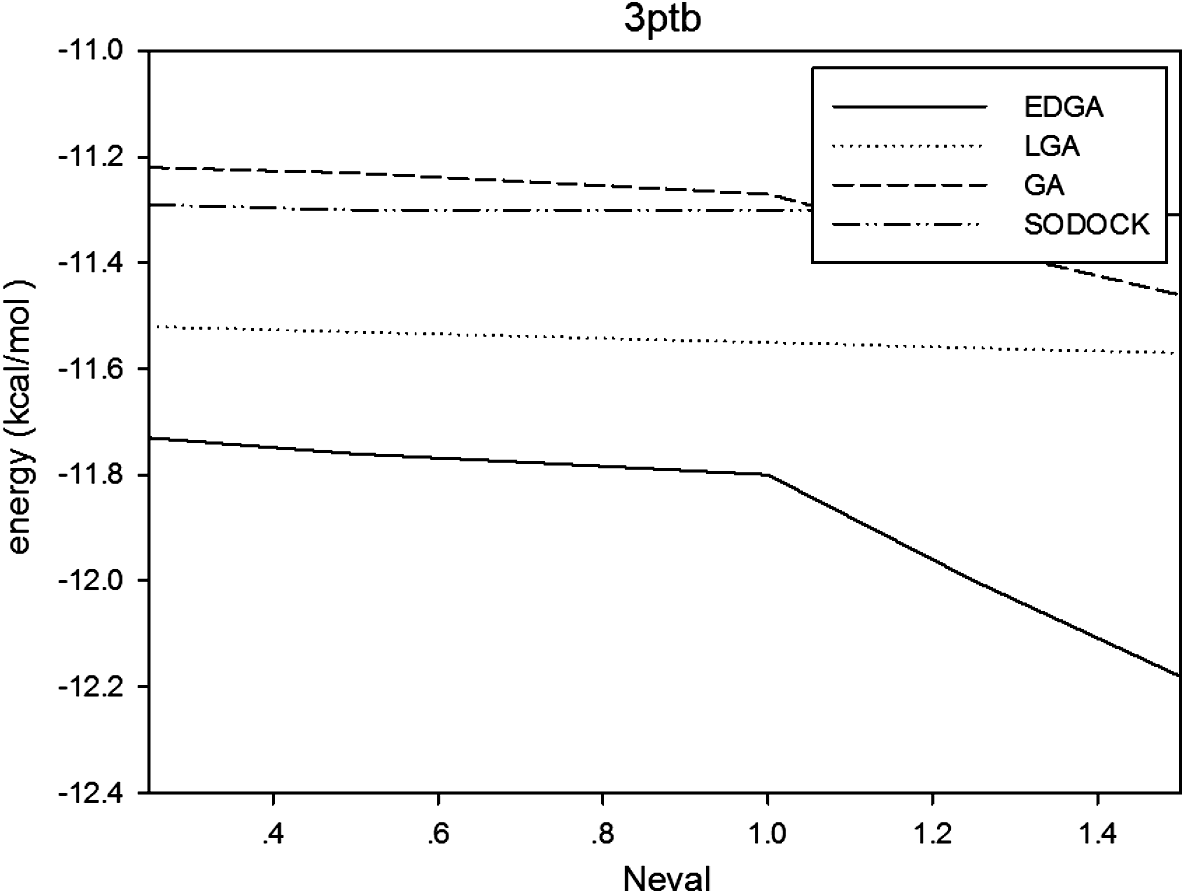
Convergence diagram of 3ptb.
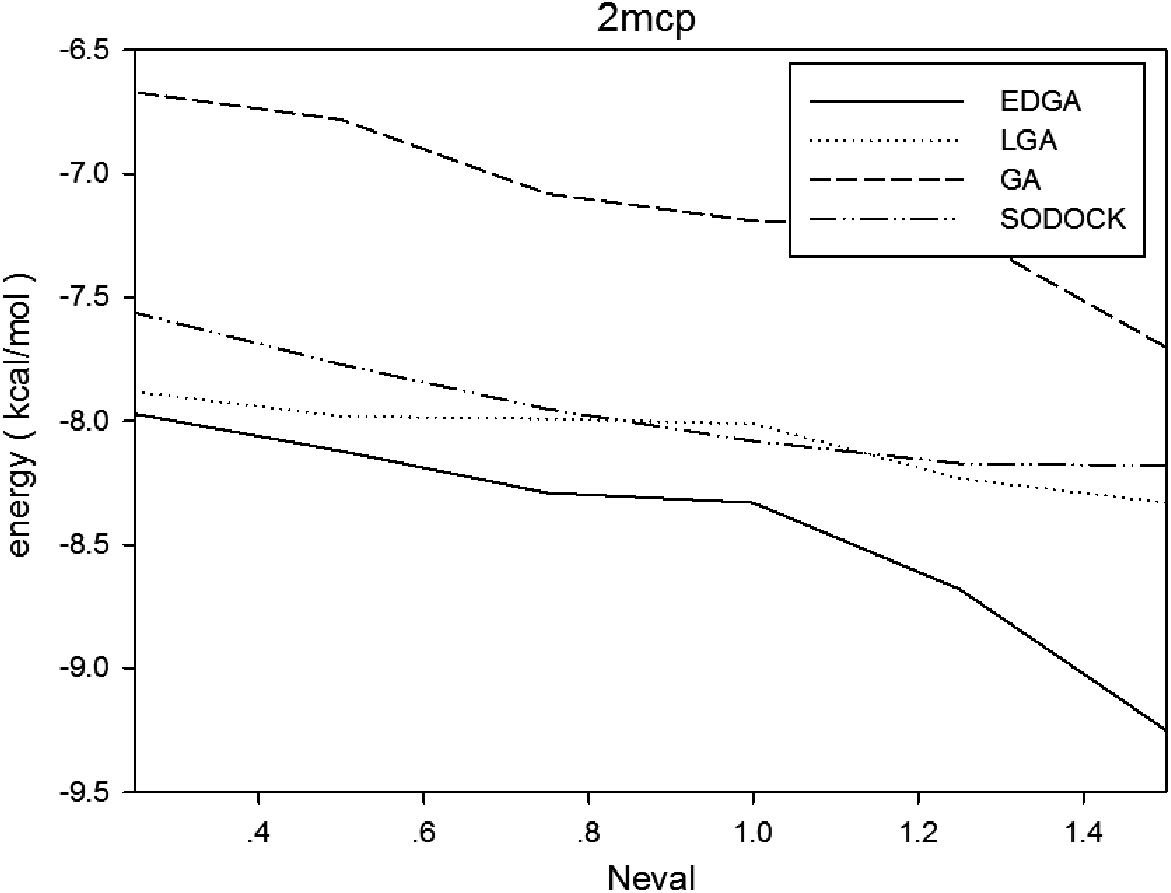
Convergence diagram of 2mcp.
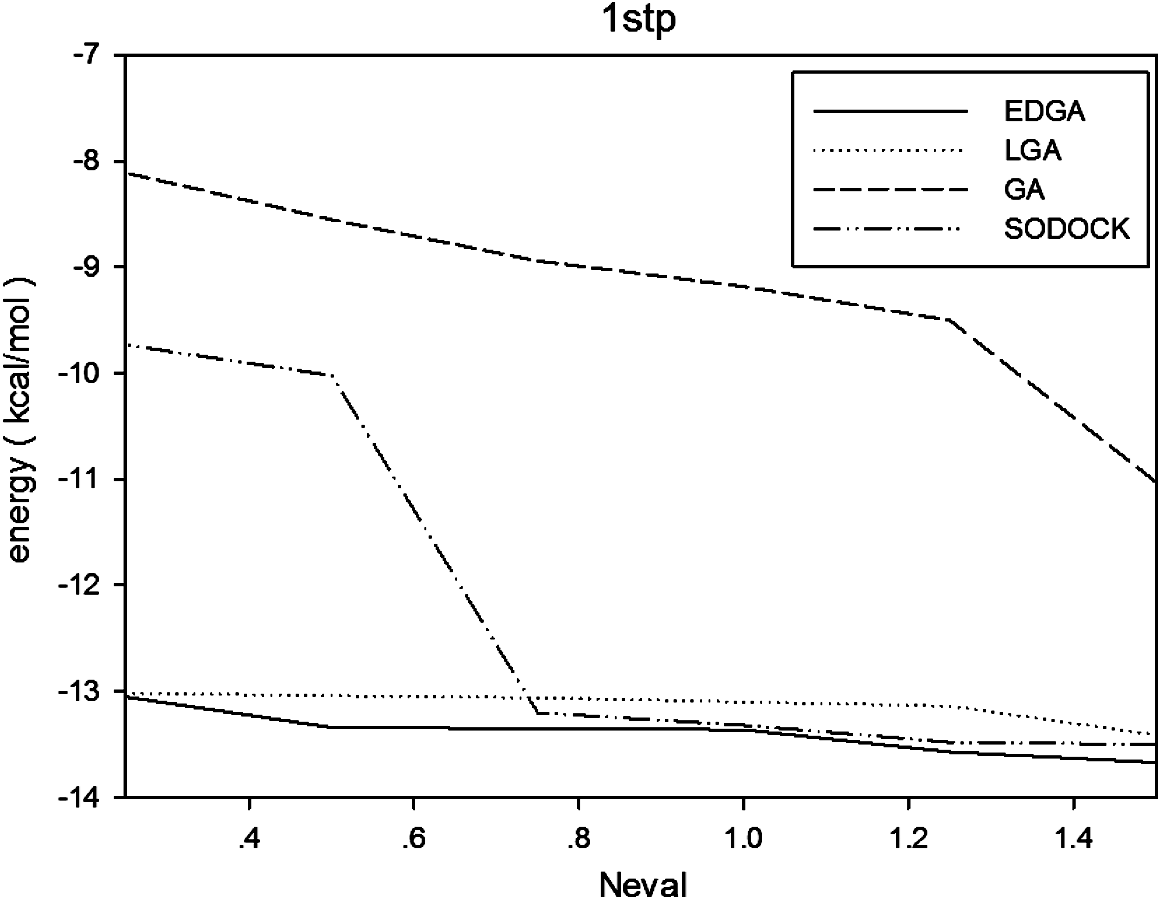
Convergence diagram of 1stp.

Convergence diagram of 1hvr.
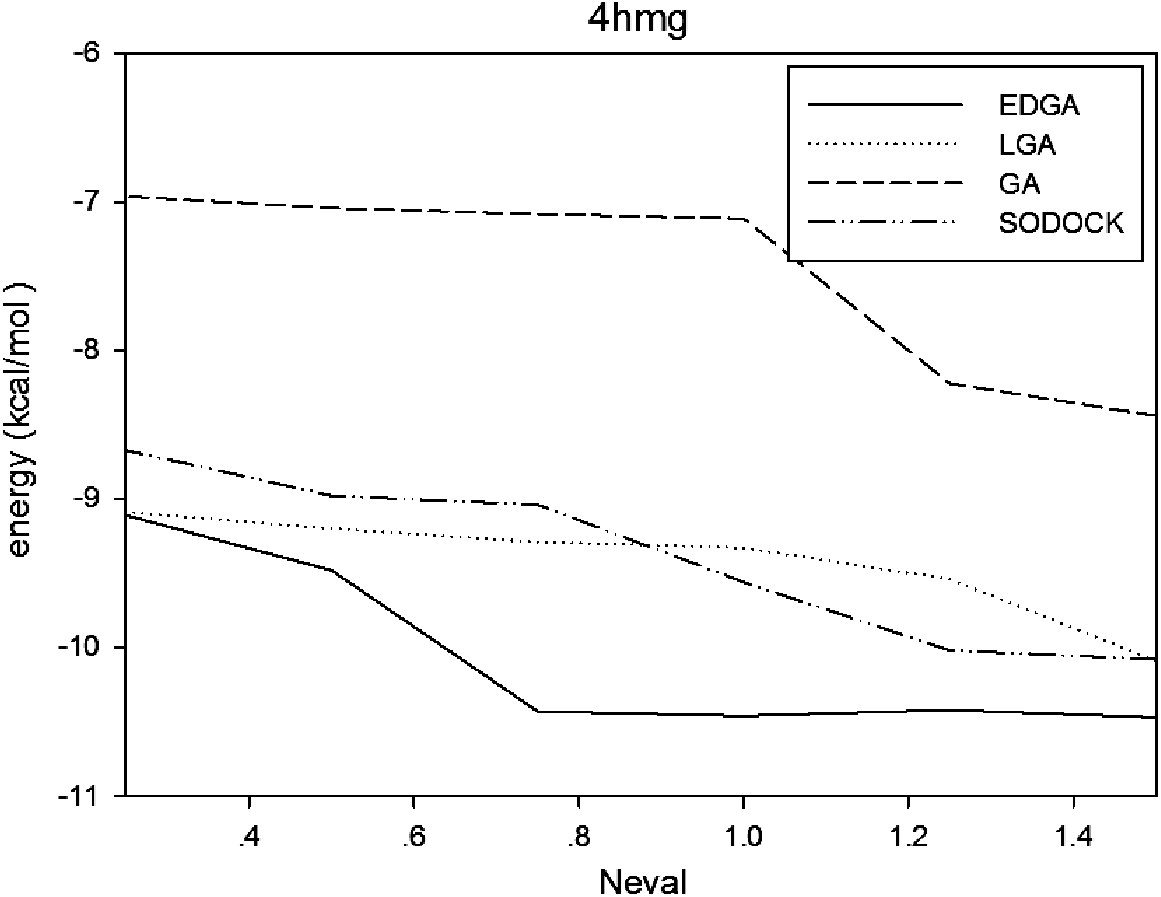
Convergence diagram of 4hmg.
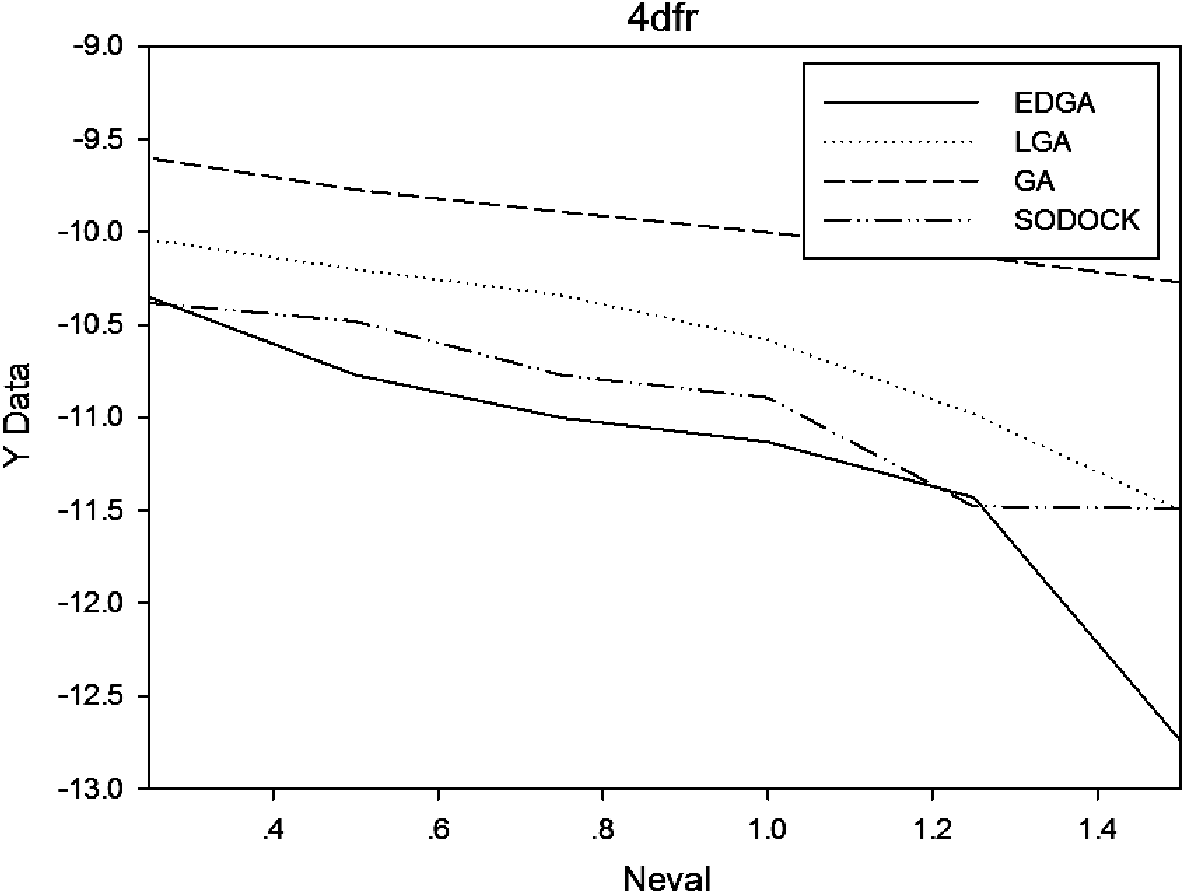
Convergence diagram of 4dfr.

Box plot of 3ptb.
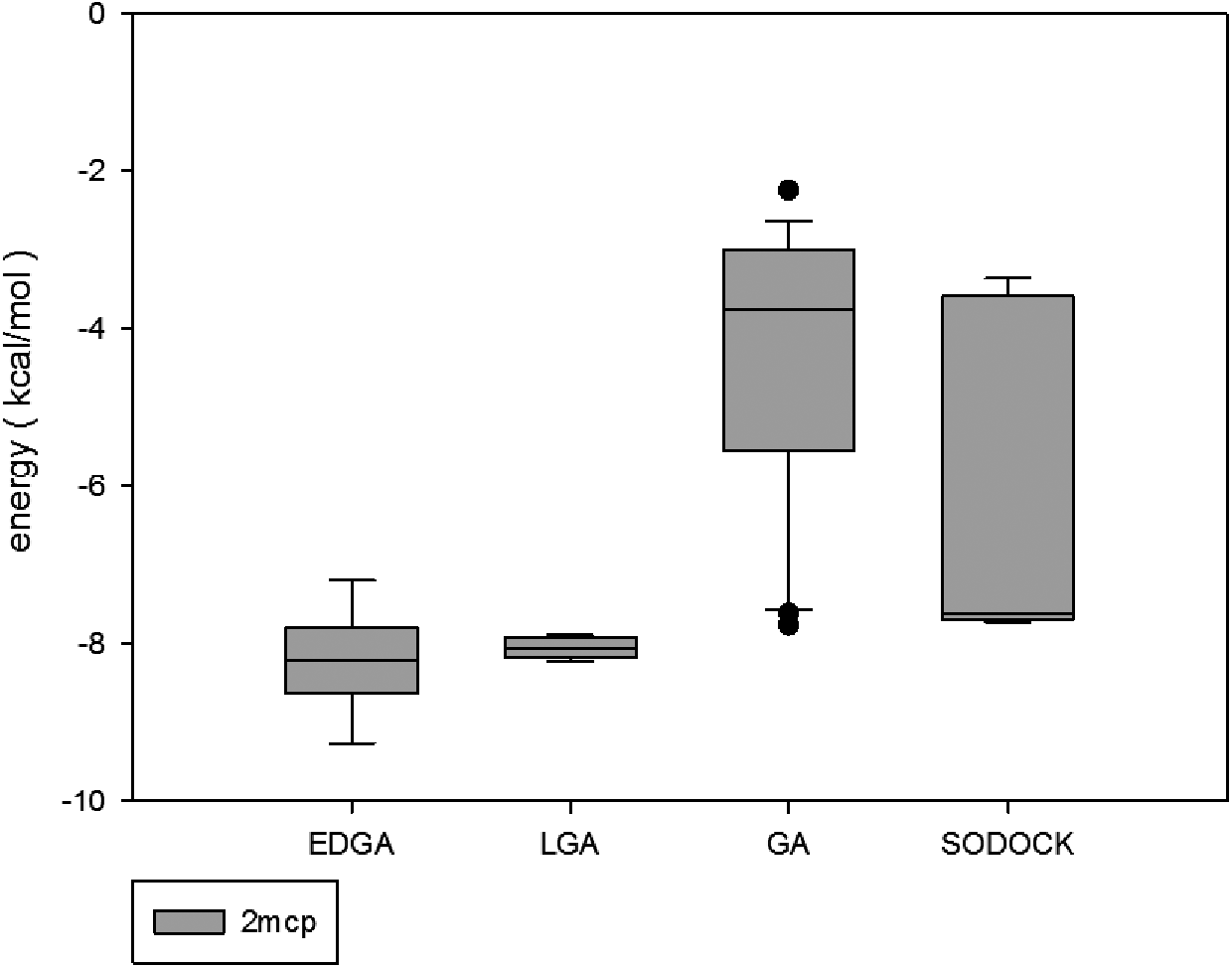
Box plot of 2mcp.
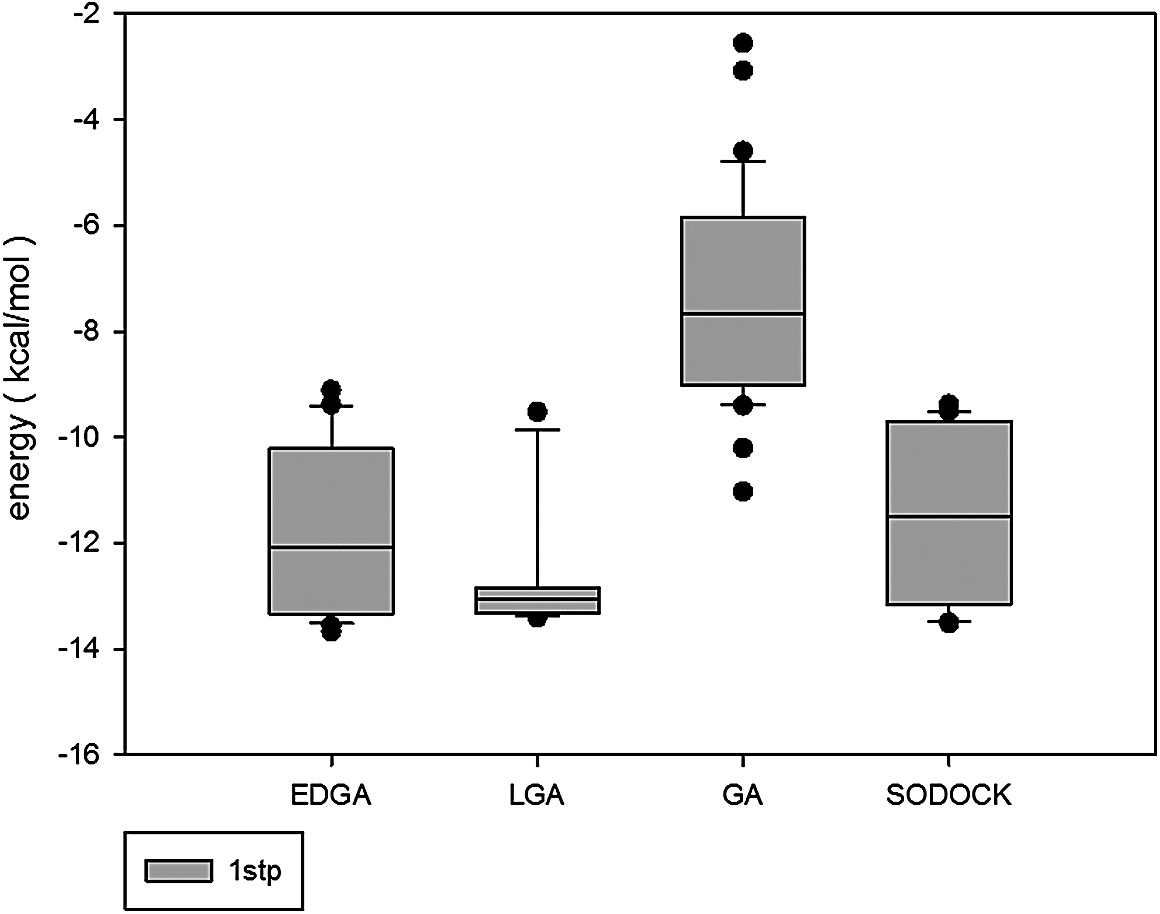
Box plot of 1stp.
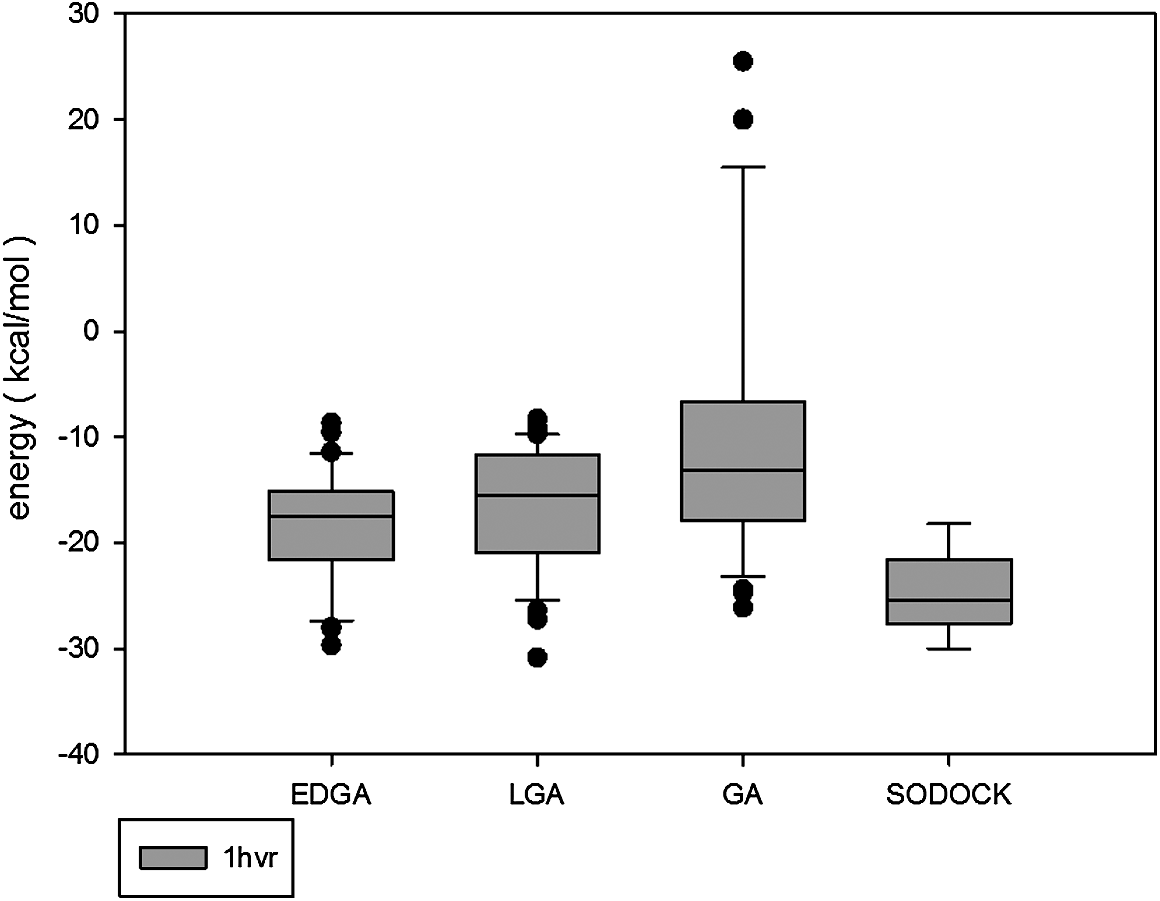
Box plot of 1hvr.
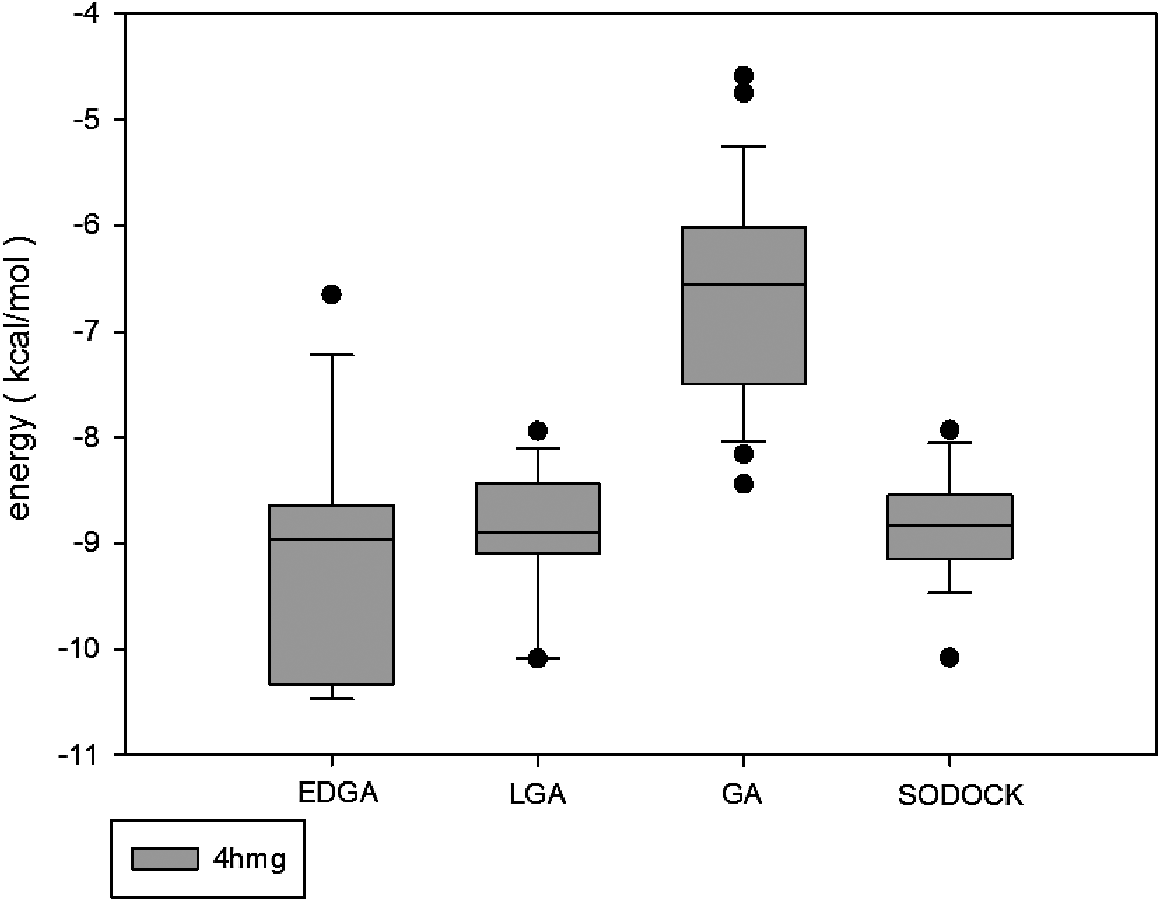
Box plot of 4hmg.

Box plot of 4dfr.
5. Conclusions
We have shown that, of the four search methods tested—GA, LGA, EDGA, and SODOCK—the most efficient, reliable, and successful is EDGA. We defined efficiency of search in terms of lowest energy found in a given number of energy evaluations, and reliability in terms of reproducibility of finding the lowest energy structure in independent dockings. The introduction of the EDGA search method extends the power and applicability of AutoDock to docking problems compared with the earlier versions of search methods.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation Program of China (61572116, 61572117, and 61502089), and the National Key Technology R&D Program of the Ministry of Science and Technology (2015BAH09F02).
Author Disclosure Statement
The authors declare that no competing financial interests exist.