
Abstract
Abstract
Noncoding ribonucleic acids (RNA) play a critical role in a wide variety of cellular processes, ranging from regulating gene expression to post-translational modification and protein synthesis. Their activity is modulated by highly dynamic exchanges between three-dimensional conformational substates, which are difficult to characterize experimentally and computationally. Here, we present an innovative, entirely kinematic computational procedure to efficiently explore the native ensemble of RNA molecules. Our procedure projects degrees of freedom onto a subspace of conformation space defined by distance constraints in the tertiary structure. The dimensionality reduction enables efficient exploration of conformational space. We show that the conformational distributions obtained with our method broadly sample the conformational landscape observed in NMR experiments. Compared to normal mode analysis-based exploration, our procedure diffuses faster through the experimental ensemble while also accessing conformational substates to greater precision. Our results suggest that conformational sampling with a highly reduced but fully atomistic representation of noncoding RNA expresses key features of their dynamic nature.
1. Introduction
N
Conformational sampling procedures based on energy evaluations, such as Monte-Carlo (Frellsen et al., 2009; Landau and Binder, 2009) and molecular dynamics (MD) (Frenkel and Smit, 2001) can accurately explore the free-energy landscape of a molecule but are computationally expensive. Sampling techniques based on motion planning (Thomas et al., 2005; Al-Bluwi et al., 2012) and loop closure using inverse kinematics (Canutescu and Dunbrack, 2003; Coutsias et al., 2004; Cortés et al., 2004; Shehu et al., 2006; Yao et al., 2008) can significantly increase the efficiency of such methods. Constraint-based samplers rely exclusively on the geometry of the molecule and nonlocal constraints (Wells et al., 2005; Zavodszky et al., 2004; Yao et al., 2012) and easily jump large energy barriers to broadly sample conformational space. Normal mode analysis (NMA) and elastic network models are also popular constraint-based sampling methods that encode nonlocal interactions as harmonic restraints (Ma, 2005; Schröder et al., 2007; Chennubhotla et al., 2005; Al-Bluwi et al., 2013). The majority of these efficient techniques have only been implemented and tested on proteins.
In this study we present an inverse kinematics technique, kino geometric sampling for RNA (KGSrna), an efficient, constraint-based sampling procedure for RNA inspired by robotics. In KGSrna, an RNA molecule is represented with rotatable bonds as degrees of freedom (DOFs) and groups of atoms as rigid bodies. Noncovalent bonds are distance constraints that create nested cycles (Fig. 1b). To avoid breaking the noncovalent bonds, conformational changes in cycles require coordination (van den Bedem et al., 2005; Yao et al., 2008, 2012; Budday et al., 2015; Pachov and van den Bedem, 2015). Closed cycles greatly reduce conformational flexibility and consequently deform the biomolecule along preferred directions in the conformational landscape. We also integrated a differentiable parameterization of ribose conformations into the kinematic model.
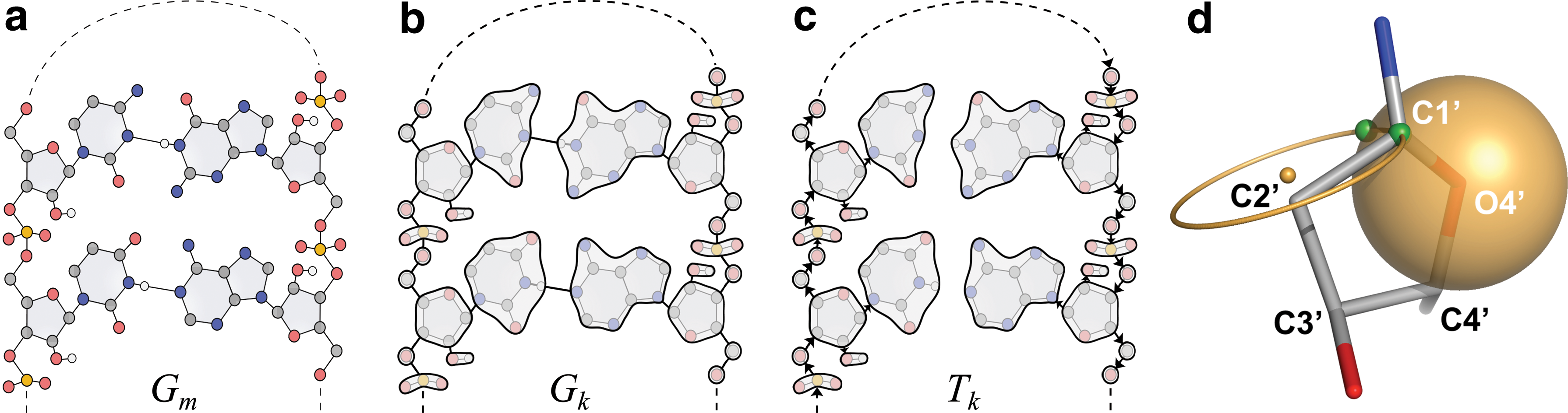
Geometric constructions: tree and ribose.
In the remainder, we first detail the methodology and the implementation of KGSrna. Next, we demonstrate that KGSrna accurately recovers all representative models in the experimental NMR bundle starting from a single member. We then perform a direct comparison with the NMA method by Lopéz-Blanco et al. (2011), which shows KGSrna maintains high quality geometry of the molecules while locally exploring more diverse portions of conformational space.
2. Methods
The purpose of KGSrna is to sample the unweighted native ensemble of RNA molecules starting from a single member of an ensemble. For this purpose, KGSrna takes as input an initial conformation,
2.1. Construction of the tree
A graph Gm = (Vm, Em) is constructed such that Vm contains all atoms and Em contains all covalent or hydrogen bonds (see Fig. 1a). In RNA, only the hydrogen bonds A(N3)–U(H3) and G(H1)–C(N3) in canonical Watson-Crick (WC) base pairs are included as edges. WC base pairs are taken as all base pairs labeled XX, XIX in the Saenger nomenclature from RNAView (Yang et al., 2003).
Next, a compressed graph Gk = (Vk, Ek) is constructed from Gm by edge contracting members of Em that correspond to (1) partial double bonds, (2) edges (u, v) where u or v has degree one, or (3) edges in pentameric rings (ribose in nucleic acids or proline in amino acids) (Fig. 1b). Each edge in Ek thus corresponds to a revolute joint, that is, a rotating bond with 1 degree of freedom, and vertices in Vk correspond to collections of atoms that form rigid bodies.
Finally, a rooted minimal spanning tree,
2.2. Modeling the conformational flexibility of pentameric rings
The flexibility of RNA is particularly dependent on conformational flexibility of ribose rings (Leontis et al., 2006), but directly perturbing a torsional angle in pentameric rings breaks the geometry of the ring. While pseudorotational angles (Altona and Sundaralingam, 1972) are frequently used to characterize ribose conformations, they are not convenient for a kinematic model as the equations mapping a pseudorotation angle to atom positions are nontrivial. We therefore introduce a parameterization inspired by Ho et al. (2005) from a continuous differentiable variable τ to the backbone δ angle, (C5′-C4′-C3′-O3′) so ideal geometry of the ribose is maintained (Fig. 1d).
The positions of O4′, C4′, and C3′ are determined by (torsional) DOFs higher in the kinematic tree. The position of C2′ and the branch leaving C3′ in the kinematic tree is determined from the C5′-C4′-C3′-O3′ torsion, δ. Thus, only the remaining atom C1′ needs to be placed. Positions of C1′ with ideal C1′-C2′ distance and C1′-C2′-C3′ angle are represented by a circle (Fig. 1d), centered on the C3′-C2′ axis and having the C3′-C2′ axis as its normal vector. Positions of C1′ that have ideal C1′-O4′ distance are represented by a sphere centered on O4′. The position of C1′ is on either of the intersections between the sphere and the circle, indicated by the variable, u
To avoid using u, which is discontinuous, and δ, which is limited by the ring geometry, we introduce the periodic and continuous variable τ, which uniquely specifies both δ and u. Since δ is restricted to move in the range 120∘ ± A where A is typically ≈40∘, we set δ = 120∘ + A cos τ. By defining u = sgn sin τ, the ribose conformation follows a continuous, differentiable, and periodic motion for τ
2.3. Null space perturbations
The full conformation of a molecule is represented as a vector
We use a constraint c
where
2.4. Rebuild perturbations
The conformation of ribose rings change when performing null space perturbations, but in general the changes are small enough that a full change from C3′-endo to C2′-endo is very rarely observed even in flexible loop regions. As shifts from one ribose conformation to another are frequent and biologically important in RNA molecules (Levitt and Warshel, 1978), a rebuild perturbation was designed that can completely change a ribose conformation and rebuild the backbone so the conformation stays on the closure manifold.
A rebuild perturbation first picks a segment of two nucleotides, neither of which are constrained by hydrogen bonds or aromatic stacking. It then disconnects the C4′-C5′ bond at the 3′ end of the segment, stores the positions of C4′ and C5′, and resamples the τ value of the two nucleotides, which breaks the C4′-C5′ bond.
To reclose the broken bond we let
In general
Ribose conformations in experimental structures mainly fall in two distinct peaks corresponding to C2′-endo and C3′-endo. To mimic this behavior, τ-angles are sampled using a mixture of wrapped normal distributions. The following bimodal distribution (Fig. 2) was obtained by fitting to the τ-angles of riboses taken from the high-resolution RNA dataset compiled by Bernauer et al. (Bernauer et al., 2011).
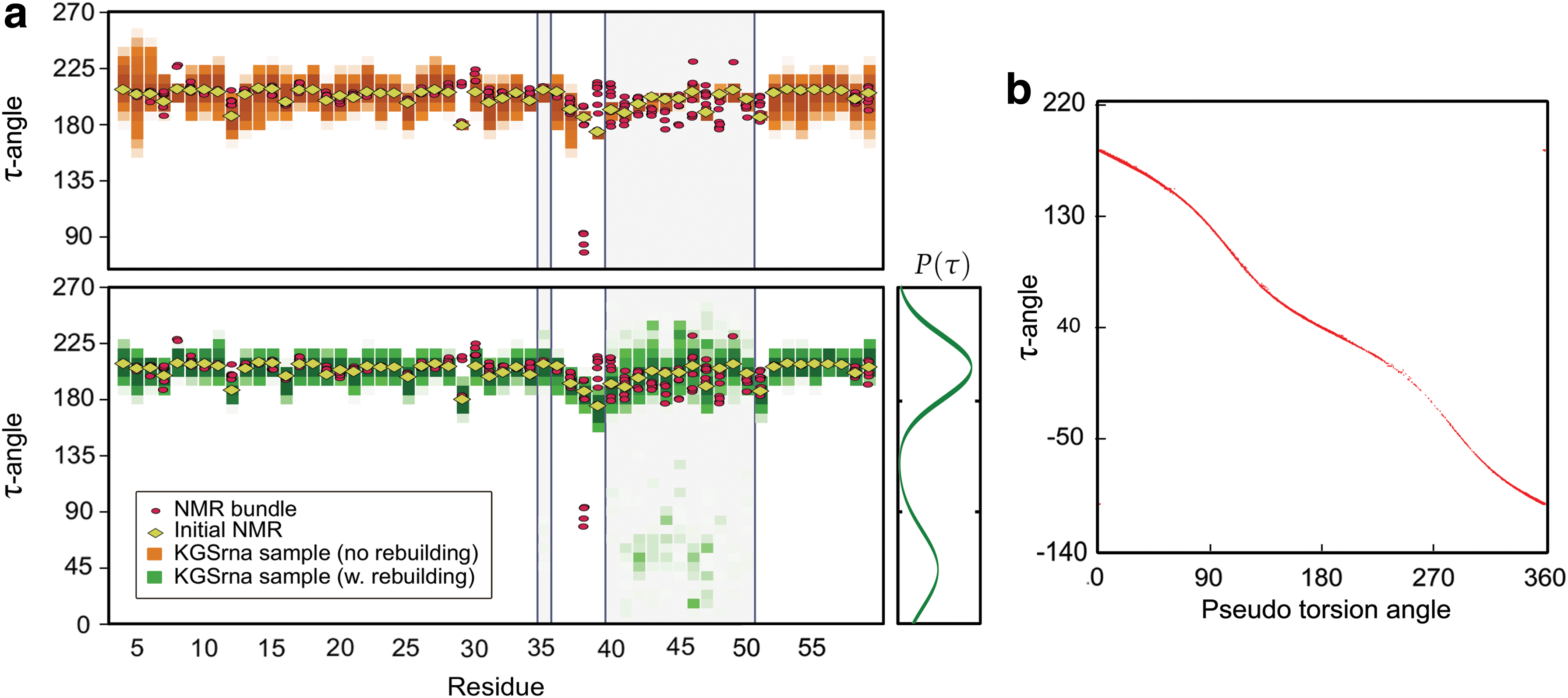
Kino geometric sampling (KGS) illustrated by τ angle.
Only nucleotides that are not part of any base-pairing or stacking, as obtained by RNAView, were included.
After resampling a loop segment most loop closure methods tend to overly distort DOFs near the endpoint of the chosen segment. Our method addresses this by (1) resampling randomly chosen segments of tow nucleotides only so the end points are not always in the same location and (2) by using the inverse Jacobian method that tends to distribute the DOF-updates more evenly along the segment than for example, cyclic coordinate descent (Canutescu and Dunbrack, 2003) or random loop generation (Cortés et al., 2004).
2.5. Experimental design
A benchmark set of sixty RNA molecules (see Supplementary Table S1, available online at www.liebertpub.com/cmb) was compiled from the Biological Magnetic Resonance Bank (BMRB) (Ulrich et al., 2008) by downloading single-chain RNAs that contain more than 15 nucleotides and are solved with NMR spectroscopy. RNAs with high sequence similarity were removed so the edit-distance between the sequences of any pair was at least five.
For each molecule in the benchmark set, the first NMR model is chosen as
If a new conformation contains a clash between two atoms it is rejected and a new seed is chosen. An efficient grid-indexing method is used for clash detection by overlapping van der Waals radii (Halperin and Overmars, 1994). The van der Waals radii were scaled by a factor of 0.5.
The iMod toolkit (Lopéz-Blanco et al., 2011) uses normal mode analysis (NMA) in internal coordinates to explore conformational flexibility of biomolecular structures, for instance via vibrational analysis, pathway analysis, and Monte-Carlo sampling. The iMod Monte-Carlo sampling application was used for comparison with KGSrna and run with the default settings: heavy-atoms, five top eigenvectors, 1000 Monte-Carlo iterations per output structure, and a temperature of 300K.
3. Results and Discussion
To assess the performance of our model in representing RNA modes of deformation, we compared the distribution of our samples to the available NMR bundles. For this purpose, we performed sampling runs that all start from a single member of the NMR bundle and diffuse out to a predefined exploration radius. We define the exploration width as the ability of KGSrna to quickly diffuse away from the starting conformation and the exploration accuracy as the ability to sample conformations close to any biologically relevant member of the native ensemble. To evaluate the width and accuracy of the exploration we consider NMR models as representative members of the native ensemble and measure how close to KGSrna samples these members are, both in terms of local measures (τ-angle distributions) and in terms of full chain measures (RMSD). KGSrna was used to generate 1,000 samples, starting from the first model of each of the sixty RNA structures in the benchmark set (Supplementary Table S1). The largest RMSD distance between any two models was used as the exploration radius for that molecule. The sampling took on average 372 seconds on an Intel Xeon E5-2670 CPU.
3.1. Broad and accurate atomic-scale sampling of the native ensemble
To assess the importance of the rebuilding procedure we evaluated the sampling with and without rebuild perturbations. Figure 2a illustrates distributions of the τ angle in KGSrna samples and NMR bundle structures for the Moloney MLV readthrough pseudoknot (PDB-id 2LC8). Without a rebuilding step, KGSrna samples show a very narrow sampling in the geometrically constrained loop region starting at nucleotide 40. With rebuilding enabled, the distributions of τ-angles widen significantly and all ribose conformations present in the NMR bundle are reproduced in the KGSrna sampling. When sampling without rebuilding, 9 out of the 196 nucleotides in the benchmark set that have both C3′-endo and C2′-endo conformations are fully recovered. When enabling rebuild perturbations all but four ribose conformations (98%) are recovered. These four are all in less common conformations, such as O4′-endo or C1′-endo. Supplementary Figure S1 shows the effects of KGSrna sampling with rebuilding on a δ/ε-plot.
Traditionally, ribose conformations are described using the pseudorotation angle, P, which depends on all five torsions in the ribose ring (Altona and Sundaralingam, 1972). Figure 2b shows the relationship between τ and P for all nucleotides in the benchmark set. While the two are not linearly related there is a monotonic relationship indicating that τ is as useful as P in characterizing ribose conformations in addition to being usable as a differentiable degree of freedom in a kinematic linkage.
3.2. Large-scale deformations
We evaluated the performance of KGSrna in probing conformational states on whole-molecule scale using the root mean square deviation (RMSD) of C4′ coordinates after optimal superposition. Figure 3a shows the evolution of the minimum and maximum distance from each of the 10 NMR bundle structures to the KGSrna sample of the Moloney MLV readthrough pseudoknot (2LC8) as the sampling progresses. The sampling has expanded to the limits of the exploration radius after 400 samples. The minimum distance to each of the noninitial NMR bundle conformations quickly converges to approximately 2Å RMSD. Both these trends are consistent across the benchmark set with an average minimum RMSD of 1.2Å as shown in Supplementary Table S1.
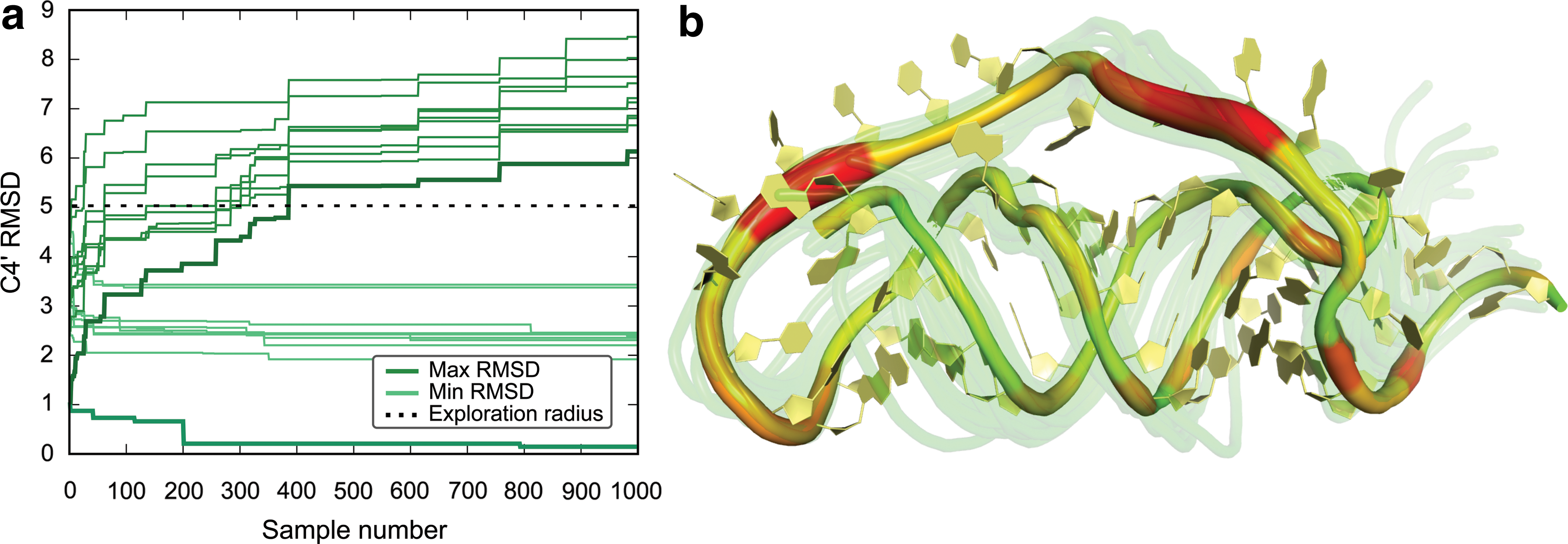
Conformational exploration of KGSrna at molecular scale illustrated using the Moloney MLV readthrough pseudoknot (2LC8).
Regions of the molecule that are either constrained by tight sterics or by hydrogen bonds are difficult to deform, which is implicitly represented in KGSrna's model of flexibility. Figure 3b uses color-coding to highlight the regions of 2LC8 where the degrees of freedom show a particularly high variance. The base-paired regions that are tightly woven in a double helix show little flexibility while the unconstrained loop region displays the highest degree of flexibility. Even though the O3′-terminal end (right-most side of Fig. 3a) does not by itself display a large degree of flexibility, it still moves over a large range as shown by the 25 randomly chosen overlaid KGSrna samples.
3.3. KGSrna as an alternative to NMA
The iMod Monte-Carlo application (iMC) is one of the state-of-the-art methods most directly comparable to KGSrna as it efficiently performs large conformational moves that reflect the major modes of deformation of biomolecules.
Figure 4a and b shows results of running iMC for 1,000 iterations on the Moloney MLV readthrough pseudoknot (2LC8). While KGSrna is able to sample sugar conformations widely, the standard deviation of τ is less than 1 degree for all nucleotides in the iMC sample set. Supplementary Figure S2 shows a similar comparison for the remaining backbone torsions. Furthermore, KGSrna samples widely and reaches the exploration radius of 5Å after 400 samples, while iMC has converged on 3.3Å after 1,000 samples. KGSrna generates structures closer than 2Å to an NMR bundle conformation while the best iMC conformation is just over 2.5Å from its nearest NMR bundle structure. This indicates both a broader exploration width and higher exploration accuracy of KGSrna compared to iMC.
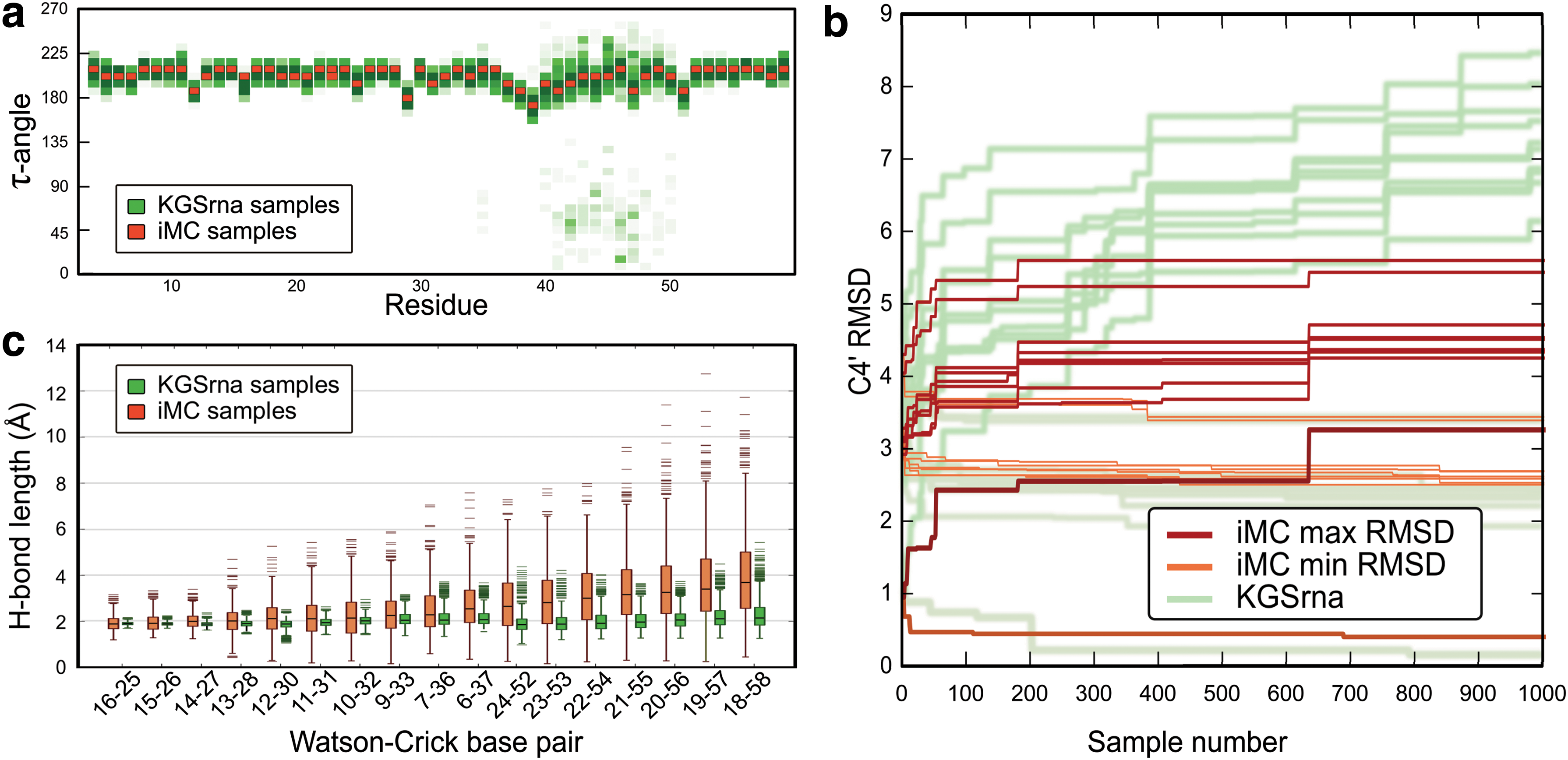
Conformational exploration of iMC illustrated using the Moloney MLV readthrough pseudoknot (2LC8).
Figure 4c shows distributions of hydrogen bond length in WC base pairs in the 1,000 samples from iMC and KGSrna respectively. The average standard deviation of hydrogen bond distances is 1.04Å for iMC base pairs, which for most applications would constitute a full break of the bond. The standard deviation is only 0.33Å for KGSrna. The source of hydrogen bond fluctuations in KGSrna is primarily the null space moves, where a relatively high step size causes the first-order approximations to introduce small deviations from the closure manifold.
An alternative approach to model the flexibility of a molecule is to predict its structure “de novo” from the sequence and secondary structure. The resulting set of “decoys” can be used as representatives of a conformational ensemble. The macromolecular conformations by symbolic programming method (MC-sym) is an RNA 3D structure modeling system that takes as input the sequence and secondary structure in dot-bracket notation (Lemieux and Major, 2002). Figure 5 shows the 175 structures generated by running MC-sym for 96 hours on the primary and secondary structure of 2LC8:
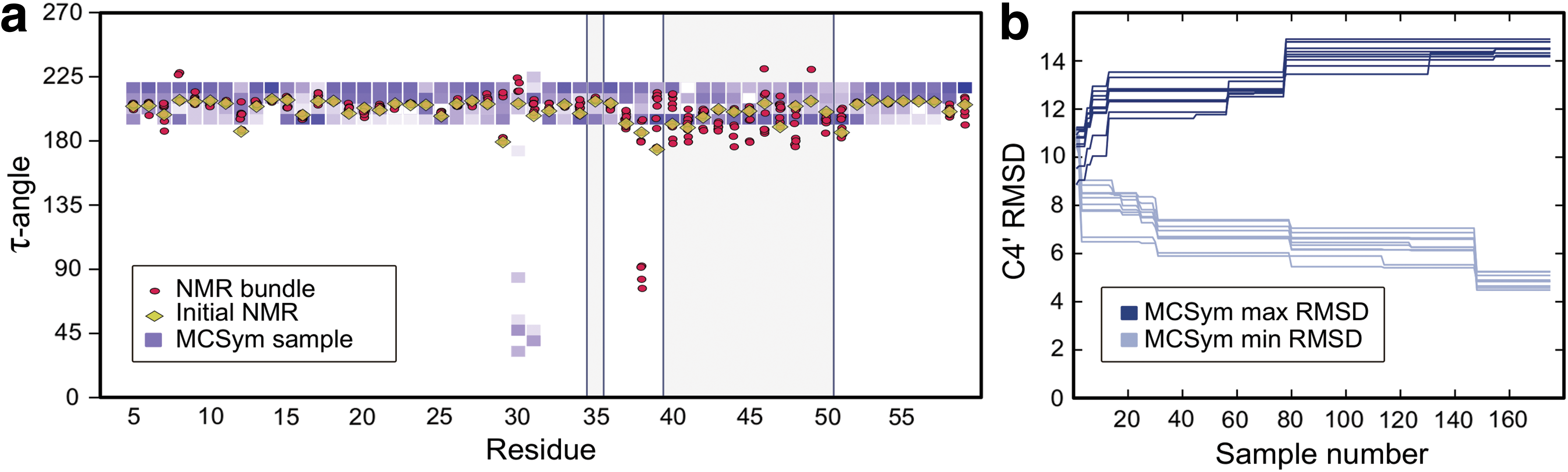
Conformational exploration of MC-sym illustrated using the Moloney MLV readthrough pseudoknot (2LC8).
GGUCAGGGUCAGGAGCCCCCCCCUGAACCCAGGAUAACCCUCAAAGUCGGGGGGCA
((((((((((((..[[[[[[[.))))..))))))))............]]]]]]].
While some flexibility is permitted for sugar conformations, they are relatively limited for most residues and fail to recover the NMR bundle conformation in the flexible loop region. The minimum RMSD to any NMR bundle member is a little under 5Å, which was the largest permitted exploration radius for KGS. This indicates that only very few of the MC-sym structures reached the native ensemble. The fragment assembly of RNA with full-atom refinement method (FARFAR) is another de novo method, but it is mainly tested on nucleic acid chains of length 20 or less. The server version rejects any chain length longer than 32 residues, which excludes a large portion of our benchmark set (Das et al., 2010; Lyskov et al., 2013).
4. Conclusion
As opposed to MD simulations, nondeterministic sampling algorithms coupled with simplified, knowledge-based potentials provide no information on dynamics but can broadly explore the conformational landscape (Bernauer et al., 2011; Das and Baker, 2007; van den Bedem and Fraser, 2015). Our analysis demonstrates that conformational ensembles of noncoding RNAs in solution are accessible from efficiently sampling coordinated changes in rotational degrees-of-freedom that preserve the hydrogen bonding network. Each member of a synthetic ensemble was approximated to within 2Å on average by a KGSrna sampled conformation on a benchmark set of sixty noncoding RNAs without relying on a forcefield. By contrast, an NMA-based sampling algorithm diffuses through the folded state at a slower rate, approximating each ensemble member with 25% less accuracy. Additionally, de novo methods that model conformational space only from primary and secondary structures were demonstrated to require much longer computation times while obtaining less sampling accuracy, which is not surprising given the more demanding nature of the problem they seek to solve.
Hydrogen bonds and similar noncovalent constraints, like hydrophobic interactions, encode preferred pathways on the conformational landscape, enabling our procedure to efficiently probe the conformational diversity resulting from equilibrium fluctuations of the ensemble. Our procedure is generic, atomically detailed, mathematically well founded, and makes minimal assumptions on the nature of atomic interactions. Combined with experimental data, it can provide insight into which substates are adopted. Our procedure is easily adapted to DNA, and protein–protein or protein–nucleic acid complexes. It could provide insights on the flexibility of interesting systems such as RNA aptamers, RNA–protein recognition, or possibly characterize riboswitch structures. Software is available online (KGS, 2016).
Footnotes
Acknowledgments
This work is part of the ITSNAP Associate Team. We thank the Inria Équipe Associée program for financial support. J.B. acknowledges access to the HPC resources of TGCC under the allocation t2013077065 made by GENCI.
This work was supported by the U.S. National Institute of General Medical Sciences Protein Structure Initiative [U54GM094586] and by a SLAC National Accelerator Laboratory LDRD (Laboratory Directed Research and Development) grant [SLAC-LDRD-0014-13-2 to H.v.d.B].
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.