
Abstract
Abstract
Genome-wide association studies have revealed individual genetic variants associated with phenotypic traits such as disease risk and gene expressions. However, detecting pairwise interaction effects of genetic variants on traits still remains a challenge due to a large number of combinations of variants (∼1011 SNP pairs in the human genome), and relatively small sample sizes (typically <104). Despite recent breakthroughs in detecting interaction effects, there are still several open problems, including: (1) how to quickly process a large number of SNP pairs, (2) how to distinguish between true signals and SNPs/SNP pairs merely correlated with true signals, (3) how to detect nonlinear associations between SNP pairs and traits given small sample sizes, and (4) how to control false positives. In this article, we present a unified framework, called SPHINX, which addresses the aforementioned challenges. We first propose a piecewise linear model for interaction detection, because it is simple enough to estimate model parameters given small sample sizes but complex enough to capture nonlinear interaction effects. Then, based on the piecewise linear model, we introduce randomized group lasso under stability selection, and a screening algorithm to address the statistical and computational challenges mentioned above. In our experiments, we first demonstrate that SPHINX achieves better power than existing methods for interaction detection under false positive control. We further applied SPHINX to late-onset Alzheimer's disease dataset, and report 16 SNPs and 17 SNP pairs associated with gene traits. We also present a highly scalable implementation of our screening algorithm, which can screen ∼118 billion candidates of associations on a 60-node cluster in <5.5 hours.
1. Introduction
A
The past several years have seen the emergence of several statistical methods that can potentially be employed to address the problems mentioned above. For the first problem of error control, Meinshausen and Bühlmann (2010) proposed a procedure known as stability selection. The insight behind this technique is that, given randomly chosen multiple subsamples, true associations of covariates (e.g., SNPs or SNP pairs) to responses (e.g., a trait) will be selected at high frequency because true association signals are likely to be insensitive to the random selection of subsamples. Second, to address the nonidentifiability problem in regression due to intercovariate correlation, a randomized lasso technique has been proposed that randomly perturbs the scale of covariates in the framework of stability selection, thereby relaxing the original requirements on small correlation for recovery of true association signals from all covariates (Meinshausen and Bühlmann, 2010). Naturally, such a scheme is expected to help distinguish between true and false associations of SNPs/SNP pairs, because only true ones are likely to be selected under the perturbations. Finally, to combat the computational challenge due to a massive number of covariates, a sure independence screening (SIS) procedure (Fan and Lv, 2008) has been proposed to contain the operational size of the regression problem under provable guarantee of retaining true signals. It is possible to use the idea behind SIS to effectively perform simple independent tests on each pair of SNPs (or individual SNPs) and discard the large fraction of candidates with no associations, such that one can end up with only O(NC) candidates (where N is sample size and C is a data dependent constant) of which no true associations will be missed with high probability. These theoretical developments notwithstanding, their promised power remains largely unleashed for practical genome-wide association mapping, especially in nontrivial scenarios such as nonadditive epistatic effects, due to several remaining hurdles, including proper models for association, algorithms for screening with such models and on a computer cluster, and proper integration of techniques for error control, identifiability, screening, etc., in such a new paradigm.
In this article, we present SPHINX [which stems from sparse piecewise linear model with high throughput screening for interaction detection(X)], a new PMR-based approach built on the advancements in statistical methodologies mentioned above. It is an integrative platform that conjoins and extends the aforementioned three components, further enhanced with techniques allowing more realistic trait association patterns to be detected. In particular, SPHINX is designed to capture SNP pairs with nonlinear interaction effects (synergistic/antagonistic epistasis) on traits using a piecewise linear model (PLM), which is better suited to model the complex interactions between a pair of SNPs and the traits. In short, SPHINX is designed as follows: using an extension of SIS based on PLM, it first selects a set of O(NC) SNPs and SNP pairs with the smallest residual sum of squares. Then it runs the randomized group lasso based on PLM on the set of SNPs and SNP pairs selected in the previous step under stability selection. Finally, it reports SNPs and SNP pairs selected by stability selection, whose coefficients are nonzero given a majority of subsamples. In Figure 1, we illustrate the overall framework of SPHINX. Note that in practical association analysis with all pairs of SNPs, we should address the three problems mentioned above simultaneously, which is a nontrivial task. To achieve this goal, we take the approach of unified framework, which requires statistically sound models and algorithms and scalable system implementations.
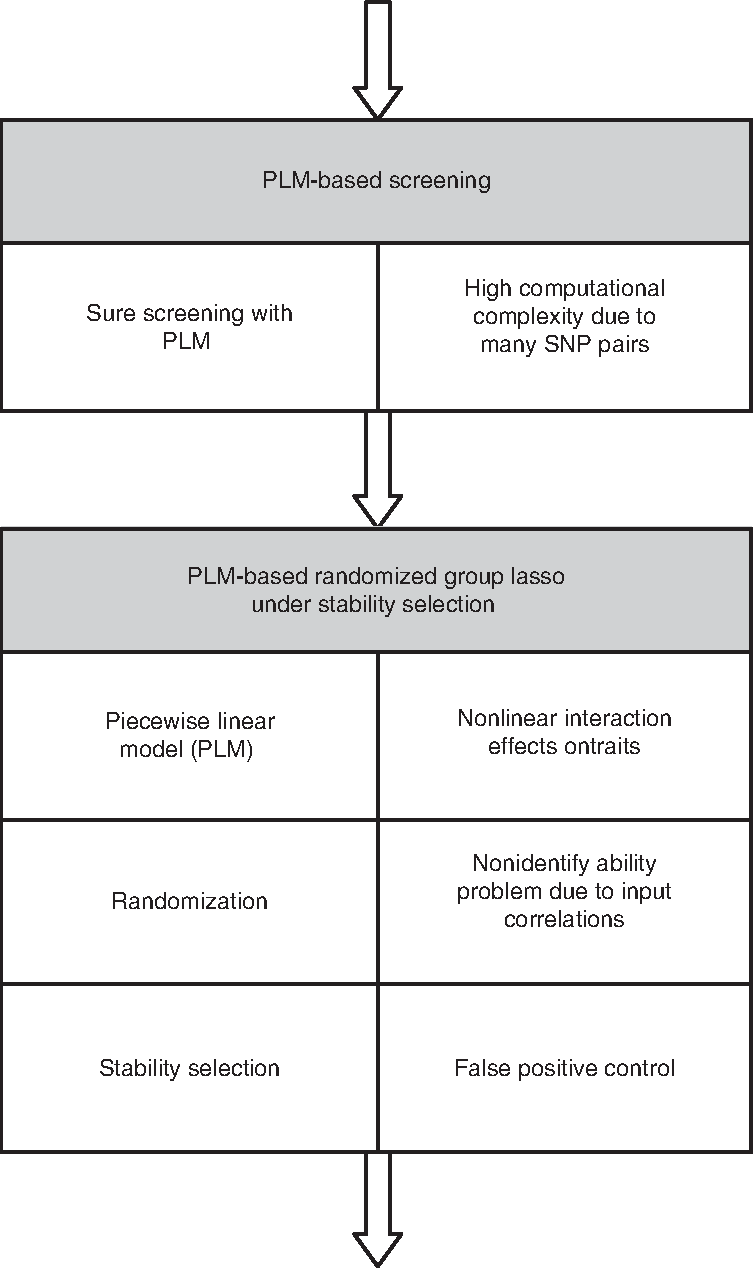
Overall framework of SPHINX. Using a screening method, we first discard SNPs/SNP pairs without associations; given that the SNPs/SNP pairs survived in the screening step, we run a method that incorporates three different techniques, each of which is introduced to address the problem on its right side.
In our experiments, we show the efficacy of SPHINX in controlling false positives, detecting true causal SNPs and SNP pairs, and using multiple cores/machines to deal with a large number of SNP pairs. Furthermore, with SPHINX, we analyzed late-onset Alzheimer's disease eQTL dataset (Zhang et al., 2013), which contains ∼118 billion candidates of associations; the analysis took <5.5 hours using 60-node cluster with 720 cores. As a result, we found 16 SNPs and 17 SNP pairs associated with gene traits. Among our findings, we report the analysis of 6 SNPs (rs1619379, rs2734986, rs1611710, rs2395175, rs3135363, rs602875) associated with immune system–related genes (i.e., HLA gene family) and an SNP pair (pair of rs4272759 and rs6081791) associated with a dopamine-related gene (i.e., DAT gene); the roles of dopamine and immune system in Alzheimer's disease have been studied in previous research (Li et al., 2004; Maggioli et al., 2013).
2. Methods
SPHINX is a framework for genome-wide association mapping, which consists of PLM-based screening technique and PLM-based randomized group lasso under stability selection. Among the SPHINX components, the effectiveness of the randomization technique and stability selection are demonstrated in Fan and Lv (2008); and Meinshausen and Bühlmann (2010) with theory and experiments; the screening approach is extensively studied in both parametric and nonparametric settings (Fan and Lv, 2008; Fan et al., 2011). In this section, we focus on describing our proposed novel model PLM-based group lasso with the randomization technique and stability selection. We then present the PLM-based screening method, followed by our system implementation of the screening method. Note that SPHINX runs the screening method prior to the PLM-based randomized group lasso, as shown in Figure 1.
2.1. Piecewise linear model-based group lasso
The relationships between genetic variations and phenotypic traits are complex, for example, nonlinear. However, due to the highly under-determined nature of the mathematical problem—too many features (SNPs and SNP pairs) but too few samples—it is difficult to employ models that have a high degree of freedom. Traditionally, linear models have been used extensively in genome-wide association studies despite the fact that these models are not flexible enough to capture the complexity of the trait-associated epistatic interactions between SNPs.
We introduce a multivariate piecewise linear model (PLM), which is better suited to model the complex interactions between a pair of SNPs and traits. Note that we employ PLM for adding additional degrees of freedom into a linear model in a high-dimensional multivariate regression setting. Therefore, it is different from the cases, in which we change the degrees of freedom in statistical tests such as the F-test. We denote the j-th SNP for the i-th individual by
where
where ujk, tjk, and wjk represent the regression coefficients for the first, second, and third line segment, respectively. Given the model in Equation. (1), to select significant SNPs/SNP pairs, we propose the following penalized multivariate piecewise-linear regression, referred to as PLM-based group lasso:
where λ1 and λ2 are regularization parameters, determining the sparsity of the solutions. Here the first ℓ1 and second ℓ1/ℓ2 norm are introduced to set the coefficients of individual SNPs and SNP pairs to exactly zero respectively if they are irrelevant to the observed trait

Here P(Wj = 1) = P(Wj = δ) = P(Wjk = 1) = P(Wjk = δ) = 0.5, and δ ∈(0, 1] determines the degree of perturbations (the smaller δ, the larger perturbations). It has been shown that this randomization with stability selection weakens the condition for the recovery of true nonzero coefficients (Meinshausen and Bühlmann, 2010). Furthermore, Meinshausen and Bühlmann empirically showed that the randomization is very useful to distinguish between true causal signals and the false ones merely correlated with the true signals (Meinshausen and Bühlmann, 2010). However, there is trade-off for the degree of random perturbations: as we increase the degree of perturbations, false positives will be reduced, but true negatives can be increased.

where q
2.2. Piecewise linear model-based screening
In Equation. (4), we include all P SNPs and
Our screening algorithm is a variant of iterative sure independence screening based on Equation. (1). It greedily selects SNPs and SNP pairs based on the contribution of each candidate SNP or SNP pair to the decrease of residual sum of squares. The PLM-based screening is described in Algorithm 2. Note that there are two pairs of parameters (kmar, kint) and (bmar, bint). The pair (kmar, kint) determines the total number of SNPs and SNP pairs selected, and (bmar, bint) determines the number of candidates selected per-iteration. In our experiments, we used (kmar, kint) = (N, N) and (bmar, bint) = (10, 10) to limit the number of selected correlated SNPs/SNP pairs at each iteration by 10. When SNPs are highly correlated, we recommend large values of (kmar, kint) and small values of (bmar, bint), because it will allow us to select more independent SNPs/SNP pairs. After the screening step, we obtain small candidate sets of SNPs and SNP pairs, and thus it is computationally tractable to solve the high-dimensional problem in Equation (4).
2.3. System implementation of piecewise linear model-based screening
We implemented a highly efficient shared- and distributed-memory parallel PLM-based screening algorithm in C++. Our implementation can exploit parallelism when running on multicore machines, or on clusters of multicore machines. To exploit shared-memory parallelism, we used PFunc (Kambadur et al., 2009), a lightweight and portable library that provides C and C++ APIs to express task parallelism. For distributed-memory parallelism, we used MPI (Message Passing Interface Forum, 1995, 1997), a popular library specification for message-passing that is used extensively in high-performance computing. In this section, we briefly describe some salient features of our implementation that optimize memory and computational efficiency.
First of all, we optimize the memory footprint of SPHINX by storing each SNP using 2 bits (to represent 0, 1, 2), thereby giving us four SNPs per-byte of data. This way, the entire SNP dataset is compressed and most of the operations, such as tests for SNP–SNP interactions, are performed as bit-wise operations. For example, using this scheme, a 200-patient, 500,000-SNP dataset only occupies 250 MB of storage that can be entirely cached in-memory on most modern machines. The SNP–SNP interaction pairs are constructed on-the-fly in order to save space instead of explicitly storing
We also optimize our implementation for computational efficiency. Note that the significant SNPs and SNP–SNP interactions are selected by solving millions of linear systems, followed by computation of the 2-norm of the resulting residual. To quickly solve the linear systems, we use the Cholesky factorization (Bretscher, 1997) because Cholesky decomposition is faster (although less numerically stable in some cases) than other alternatives such as QR decomposition (Bretscher, 1997) and singular value decomposition (SVD) (Golub and Reinsch, 1970). Furthermore, we use BLAS and LAPACK kernels to optimize all the linear operations. After the selection of the first SNP and SNP pair, incremental linear models are built; that is, given a set of selected SNPs and SNP pairs, the best SNP or SNP pair to add to our model has to be determined. As the number of selected candidates increases, the linear system becomes more expensive to solve, thereby making successive later iterations expensive. In order to offset these costs, we resort to using a hand-coded incremental version of the Cholesky factorization, which keeps the per-iteration costs near constant.
3. Simulation Study
In this section, we validate the effectiveness of SPHINX in terms of false positive control, statistical power, and the benefits of using a piecewise linear model over a linear model via simulations because ground-truth associations are unknown in real datasets. Furthermore, we show the scalability of our screening implementation on multinode, multicore, and hybrid settings. We first conduct an extensive simulation study to demonstrate and statistically validate the efficacy of SPHINX, in comparison to two popular existing approaches: the two-locus test by PLINK (Purcell et al., 2007) with the –epistasis option and the maximum likelihood method with the fully parametrized two-locus model (saturated two-locus test) (Evans et al., 2006) with Bonferroni correction at significance level 0.01. We set |Ωmar| = N, |Ωint| = N, and κ = 3 for SPHINX, allowing for three false positives on average, and used the following sequence of regularization parameters: λ1 = λ2
3.1. Generation of simulation data
Let us denote
where yi is the continuous response (e.g., gene expression level) for the i-th individual,
For the nonadditive scenario, we generate simulation data as follows:
where
where rq ∼ Unif(−βjk,βjk) for all q = 1,…, 4. Note that in this nonadditive scenario, the relationship between
In our experiments below, we denote N by the sample size, P by the number of SNPs,
3.2. False positive control
We first confirm that SPHINX effectively controls the number of false positives of SNP pairs under two null hypotheses: (1) there exist no marginal and no interaction effects, and (2) there exist only marginal effects but no interaction effects. As shown in Figure 2, for both null hypotheses, false positives were well controlled with different sample sizes from 100 to 1000 and different numbers of SNPs from 100 to 700 (less than one false positive under both null hypotheses). However, PLINK and the saturated two-locus test did not effectively control the number of false positives under the second scenario (up to 7.94 and 8470, respectively) because SNP pairs correlated with SNPs having some marginal effects were falsely detected.
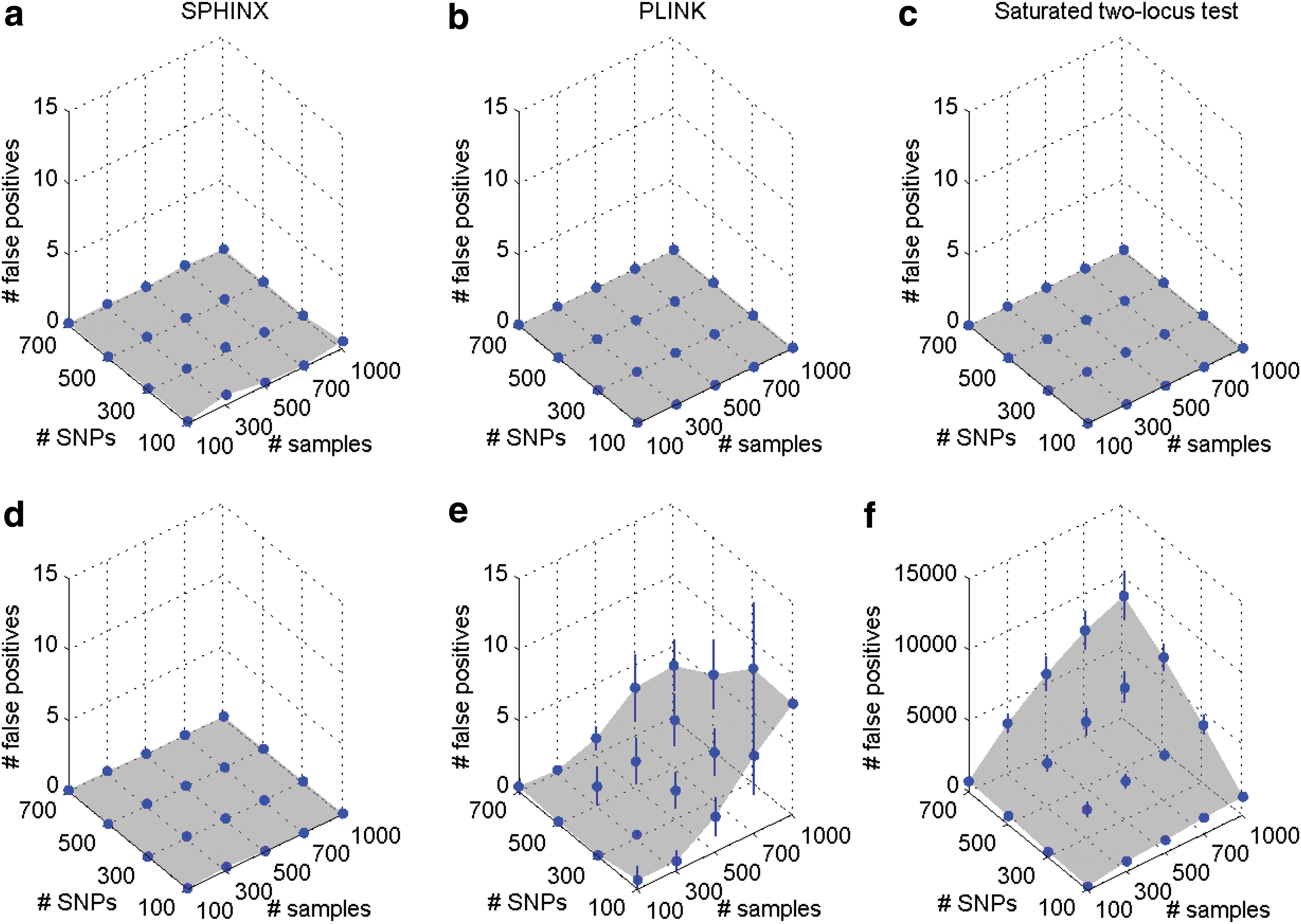
Number of false positives of SNP pairs found by SPHINX
3.3. Comparison of different methods for the detection of SNP pairs with interaction effects
We present our comparison results among SPHINX, the two-locus test by PLINK (Purcell et al., 2007) with the –epistasis option, and the saturated two-locus test (Evans et al., 2006) with various experimental settings. We evaluate the performance of SPHINX, PLINK (Purcell et al., 2007), and the saturated two-locus test (Evans et al., 2006) on simulation datasets with different numbers of true association SNP pairs, different MAFs of true association SNP pairs, and different association strengths.

Comparison of true positive rate and the number of false positives among SPHINX, PLINK, and saturated two-locus test with different numbers of true association SNP pairs under the additive scenario
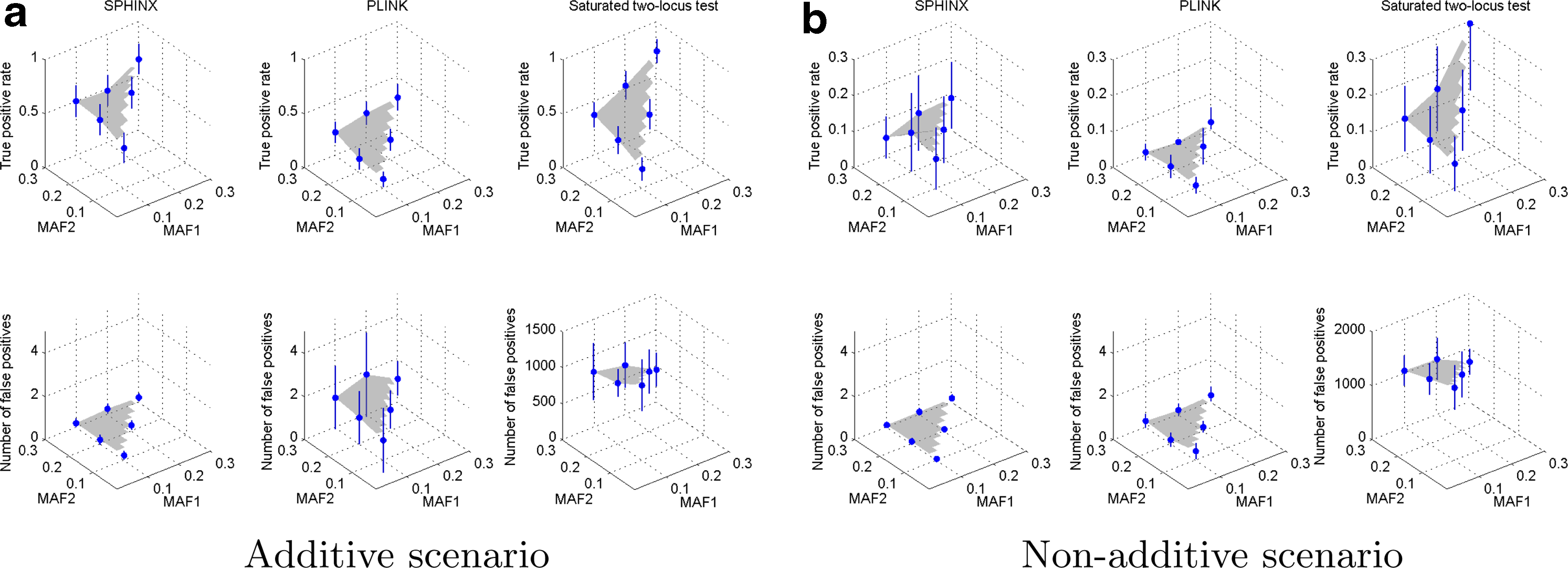
Comparison of true positive rate and the number of false positives for SPHINX (first column), two-locus test by PLINK (second column), and saturated two-locus test (third column) under the linear scenario
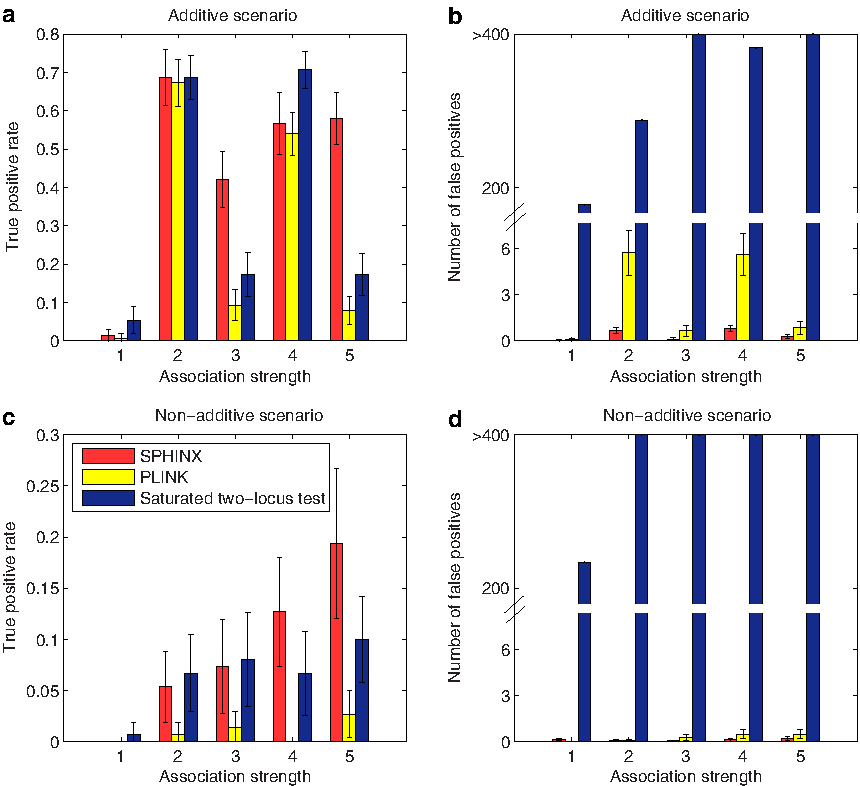
Comparison of true positive rate and the number of false positives among SPHINX, PLINK, and saturated two-locus test with different association strengths under the additive scenario
3.4. Benefits of using a piecewise linear model for screening
We tested the benefits of using a piecewise linear model instead of a simple linear model during the screening procedure. Throughout this section, we use PLS to indicate using a piecewise linear model for screening and LS to indicate using a simple linear model for screening. For this experiment, we simulated data with P = 500 (that generates candidates of 124750 SNP pairs), three SNPs having marginal effects with association strength of 1, and three SNP pairs having interaction effects with association strength of 3. Given the simulation data, for both PLS and LS, N candidates of SNP pairs were selected. We then evaluated true positive rate of PLS and LS under different minor allele frequencies (MAFs) of true association SNP pairs from 0.1 to 0.4 (fixing N = 200) and different sample sizes from 100 to 600 (fixing MAF1 = MAF2 = 0.1). Figure 6 represents the average true positive rate of PLS and LS with error bars of 1/2 standard deviation when the underlying true interaction effect was additive (Fig. 6a and b), and nonadditive (Fig. 6c and d). In general, our results show that PLS is very useful under various simulation settings. When true model was linear, true positive rates of PLS and LS were comparable in most of our settings as shown in Fig. 6 a,b), which was not expected because a simple linear model would be ideal given finite data under the additive scenario. It seems that the model complexity of PLS was small enough not to lose much power. When true model was nonlinear, PLS showed clear benefits over LS. As seen in Figure 6c and d, the true positive rate of PLS substantially increased as sample size and minor allele frequency increased but the true positive rate of LS marginally improved. It seems that the true positive rate of PLS significantly increased due to the fact that additional degrees of freedom allowed PLS to fit well into the data under the nonadditive scenario.

Comparison of true positive rate between piecewise linear screening and linear screening under different sample sizes and MAFs of true association SNP pairs under additive scenario
3.5. Scalability of Screening Implementation
We carried out scalability experiments for our screening implementation on oxygen, a six-node cluster of dual-socket, quad-core Intel Xeon E5410 machine with 32GB of RAM per-node running Linux Kernel 2.6.31-23 (total 48 cores). Figure 7 shows the throughput (SNPs processed per second) for various scenarios when running on a simulated dataset that had 200 SNPs, 500 samples, and 20 true association SNPs. Each experiment was run for 50 iterations, where each iteration considered 200 marginal candidates and
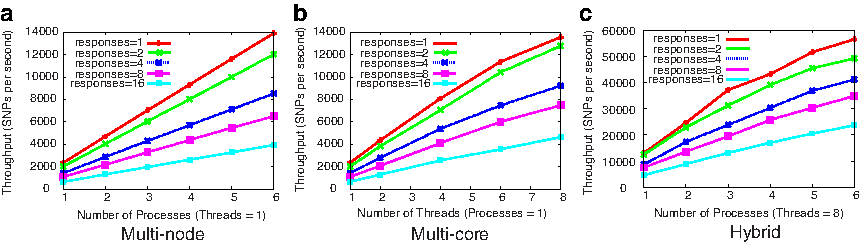
Performance of the parallel implementation of our screening algorithm on oxygen cluster.
4. Association Analysis of Late-Onset Alzheimer's Disease Data
We applied SPHINX to late-onset Alzheimer's disease (AD) data from Harvard Brain Tissue Resource Center and Merck Research Laboratories (Zhang et al., 2013) in an attempt to detect causal SNPs associated either marginally or epistatically to molecular traits of interest. This data concerns 206 AD cases with 555,091 SNPs in total and expression levels of 37,585 DNA probes including known and predicted genes, miRNAs, and noncoding RNAs in three brain regions including cerebellum, visual cortex, and dorsolateral prefrontal cortex, profiled on a custom-made Agilent 44K microarray. Specifically, we are interested in the expression traits of all 718 genes in visual cortex related to neurological diseases according to GAD (genetic association database) (Becker et al., 2004), and we focused on the 18,137 SNPs residing within 50 kb from these genes, in an attempt to search for cis-acting causal SNPs or “restricted” trans-acting (i.e., acting on genes within the same functional group) SNPs related to neurological diseases. This results in a massive problem involving 18,137 SNPs, ∼164 million SNP-pairs, and 718 gene traits, that is, ∼118 billion candidates of associations between SNPs/SNP-pairs and traits. We employed a cluster with a total of 720 cores (see Methods for experimental details), which took 4.5 hours to perform screening and <1 hour for stability selection with PLM-based randomized group lasso. Using SPHINX, we found 16 SNPs and 17 SNP pairs significantly associated with the expression traits (see Tables 1 and 2 for the list of all SNPs and SNP pairs found by SPHINX). Note that most association studies on AD have focused on detecting SNPs with marginal effects, and SNP pairs associated with AD are largely unknown. The patterns of marginal and interaction effects are illustrated in Figure 8.
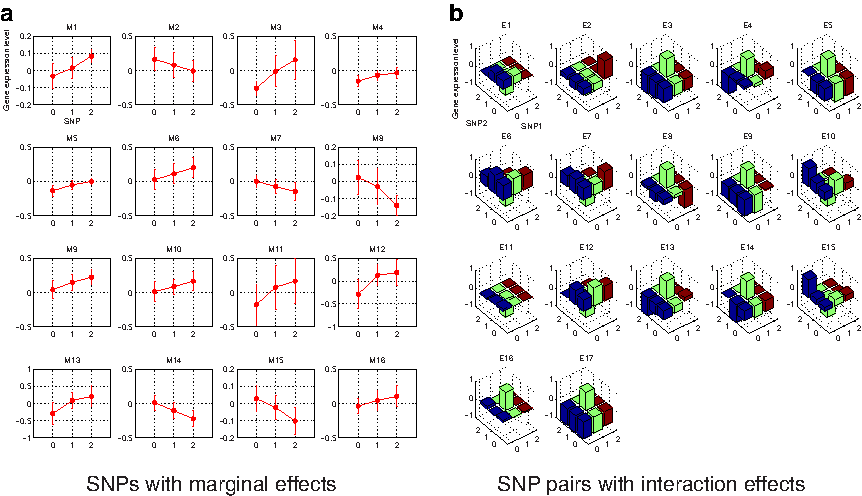
Gene expression levels according to the genotypes of
For each SNP, we represent GENE, which is located within 50 kb from the corresponding SNP. The stability score represents the proportion for which the SNP was selected in stability selection.
For each SNP A(B), we represent GENE A(B), which is located within 50 kb from the SNP. The stability score represents the proportion for which the pair was selected in the stability selection.
4.1. Marginal effects in late-onset Alzheimer's disease dataset
Among 16 SNPs identified with marginal effects, 13 SNPs were located near affected genes (12 SNPs are located within 50 kb, and 1 SNP is located within 130 kb from their associated genes), and 3 SNPs were associated with a gene trait in a different chromosome. As an example, here we investigate 6 SNPs (rs1619379, rs2734986, rs1611710, rs2395175, rs3135363, rs602875) associated with HLA (human leukocyte antigen) genes including HLA-A, HLA-DRB1, and HLA-DQB1, related to the immune system. All 6 SNPs were located nearby the affected HLA genes, which encode proteins for antigen presentation (Bodmer and Bodmer, 1978). We observed that 5 SNPs out of the 6 SNPs had positive correlation with the expression levels of their associated genes, whereas 1 SNP (rs1619379) had negative correlation with the expression levels of its associated gene (HLA-A).
We found out that 5 SNPs (out of the 6 SNPs) had genome annotations in their locations. For associations between the three SNPs (rs1619379, rs2734986, rs1611710) and HLA-A, we observed that rs1619379 and rs1611710 coincide with H3K27Ac histone mark and transcription factor binding sites, respectively, and rs2734986 aligns with spliced ESTs. It suggests that rs1619379 and rs1611710 may perturb regulatory elements of HLA-A, and rs2734986 may be related to a mechanism for DNA transcription. In case of the association between rs2395175 and HLA-DRB1, rs2395175 was in an intron of a HLA-DRB1 gene (chr6:32489683-32557613). Finally, for associations between the two SNPs (rs3135363, rs602875) and HLA-DQB1, we observed that rs3135363 coincides with both transcription factor binding site and H3K27Ac histone mark, which hints that rs3135363 may be related to regulatory mechanisms for HLA-DQB1. For rs602875, we did not find any specific genome annotations.
It should be noted that associations between HLA genes and late-onset AD (Lehmann et al., 2001; Maggioli et al., 2013) have been found, and these findings have been replicated in previous studies. It has been reported that there is association between HLA-A and late-onset of AD (Payami et al., 1997; Guerini et al., 2009), and Lehmann et al. (2006) replicated the association between HLA-B7 and AD. Furthermore, recently HLA-DRB1 identified by SPHINX has been reported as a new susceptibility locus for AD (Lambert et al., 2013). Lambert et al. identified 11 new loci associated with AD that includes HLA-DRB1 from 17,008 AD cases and 37,154 controls. This dataset is independent from ours, which indicates that our findings can be reproducible. As we found associations between the 6 SNPs and HLA genes, and previous studies reported associations between HLA genes and AD, it would be interesting to further investigate whether these associations are related to regulatory mechanisms or transcription factor bindings.
4.2. Interaction effects in late-onset alzheimer's disease dataset
Among 17 SNP pairs identified with interaction effects, as an example, we investigate the biological underpinnings of one of our findings—the pair rs4272759 (chr11:100899750) and rs6081791 (chr20:1988298) that is jointly (but not marginally) associated with DAT (dopamine active transporter, chr5:1392905-1445545) — to demonstrate the biological validity of our results. Specifically, the expression level of DAT is high only when both SNPs are heterozygous (i.e., both SNPs have only one minor allele). SNP rs4272759 is located 605 base-pairs upstream of the start position of gene PGR (progesterone receptor, chr11:100900355-101001255), whereas SNP rs6081791 is 13,407 base-pairs downstream of gene PDYN (prodynorphin, chr20:1959402-1974891). An extensive literature survey has yielded intriguing biological evidence to explain the association involving DAT with PGR and PDYN, suggesting that our finding is biologically plausible. It was reported that progesterone treatment could increase the dynorphin concentration and prodynorphin mRNA level (prodynorphin is the precursor protein of dynorphin) (Foradori et al., 2005), suggesting that a disruption of the PGR function could alter the activity of PDYN, which supports our finding that the SNPs in PGR and PDYN are epistatic. A direct association between PDYN and DAT has also been reported. For example, it has been reported that prodynorphin expression in the striatum is associated with D1 dopamine receptor stimulation (Gerfen et al., 1990); furthermore, in the experiments with DAT knock-down mice, Cagniard et al. (2005) found that the increased level of dopamine is associated with the level of dynorphin expression. Overall, evidence from the literature seems to support a hypothesis drawn from our association analysis of interacting genetic variations that a pair of SNPs affecting PGR and PDYN are likely to lead to an epistatic effect on DAT, and it would be interesting to further examine the status of DAT in the case studied in Foradori et al. (2005), and the status of PGR in cases studied in Gerfen et al. (1990) and Cagniard et al. (2005) to directly confirm and characterize such an epistatic effect.
5. Conclusions
We developed a unified framework for detecting marginal and pairwise interaction effects on traits, built on state-of-the-art techniques including screening, randomization, and stability selection. Furthermore, to facilitate the detection of SNPs and SNP pairs associated with traits at a whole genome scale, we implemented an efficient and scalable screening program. We validate the efficacy of SPHINX via simulations and the analysis of late-onset Alzheimer's disease dataset. Note that detecting pairwise interaction effects on traits requires us to address computational and statistical challenges simultaneously, which stem from a large number of SNP pairs to be tested, correlations between SNPs/SNP pairs, and nonlinear patterns of marginal and interaction effects; to our knowledge, SPHINX is the first attempt to address these challenges within a single framework. We further note that by redefining mijk in Equation (1), it is possible to investigate different choices of interaction encodings [e.g., data-driven encoding (He et al., 2015)]. In this article, we adopted the widely used genotype encoding for the pairwise interaction (i.e., multiplication of two SNPs). For future work, we plan to (1) incorporate diverse prior knowledge into our model such as trait networks using graph-guided fused lasso (Kim and Xing, 2009) or grouping information on both genotypes (e.g., LD structures) and phenotypic traits (e.g., pathways) using structured input–output lasso (Lee and Xing, 2012), (2) use kernel techniques for detecting multiway interactions among SNPs, (3) detect interaction effects under case-control settings via logistic regression, and (4) combine linear mixed model with SPHINX to correct for population structures (Rakitsch et al., 2013).
Footnotes
Acknowledgments
This work was done under a support from NIH 1 R01 GM087694-01; NIH 1RC2HL101487-01 (ARRA); AFOSR FA9550010247; ONR N0001140910758; NSF Career DBI-0546594; NSF IIS-0713379; P30 DA035778A1; and Alfred P. Sloan Fellowship awarded to E.P.X.
Author Disclosure Statement
No competing financial interests exist.