
Abstract
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR) are structured regions in bacterial and archaeal genomes, which are part of an adaptive immune system against phages. CRISPRs are important for many microbial studies and are playing an essential role in current gene editing techniques. As such, they attract substantial research interest. The exponential growth in the amount of bacterial sequence data in recent years enables the exploration of CRISPR loci in more and more species. Most of the automated tools that detect CRISPR loci rely on fully assembled genomes. However, many assemblers do not handle repetitive regions successfully. The first tool to work directly on raw sequence data is Crass, which requires reads that are long enough to contain two copies of the same repeat. We present a method to identify CRISPR repeats from raw sequence data of short reads. The algorithm is based on an observation differentiating CRISPR repeats from other types of repeats, and it involves a series of partial constructions of the overlap graph. This enables us to avoid many of the difficulties that assemblers face, as we merely aim to identify the repeats that belong to CRISPR loci. A preliminary implementation of the algorithm shows good results and detects CRISPR repeats in cases where other existing tools fail to do so.
1. Introduction
C
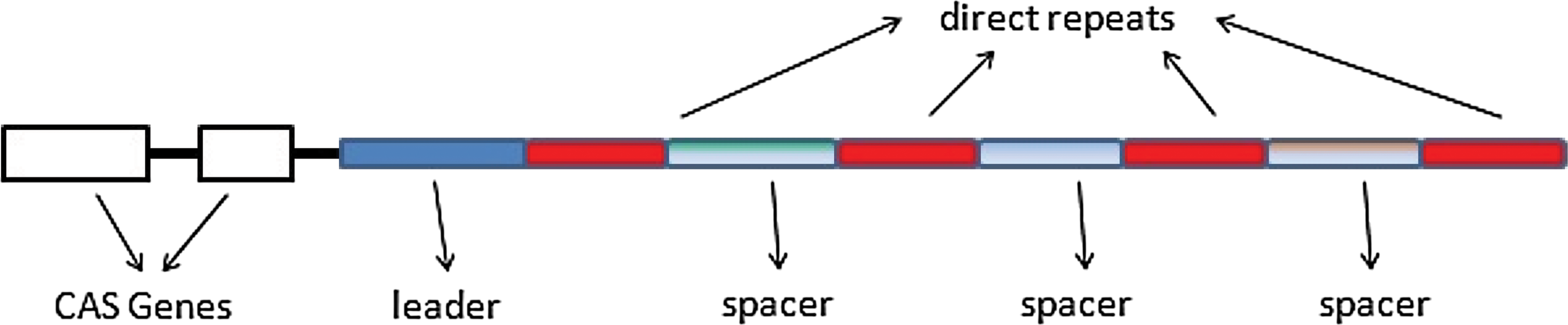
Illustration of a CRISPR locus.
CRISPRs were discovered in 1987 (Ishino et al., 1987), but their functionality was initially unknown. In 2005, it was found that spacers are derived from plasmid or phage DNA, and it was suggested that they are part of an immune system against infection by phages (Mojica et al., 2005). Today, it is known that specific Cas enzymes, guided by associated spacers, target and degrade invading foreign genetic material to stop the destruction of the host cell (Horvath and Barrangou, 2010).
New spacers are added to a locus at the 5′ end of the CRISPR array, adjacent to a leader sequence. They are inserted along with an additional repeat instance. As a consequence, CRISPR loci reflect the history of phage infections of the individual bacteria (or archaea). This fact casts new light on interesting communities, such as the bacterial community in the human gut (Stern et al., 2012). The data gathered on this bacterial community is directly related to various aspects of human health issues.
Understanding the mechanism of CRISPR and cas genes has led to an exciting research in CRISPR-Cas mediated genome editing, a CRISPR-derived novel biotechnology. This technology is playing a major role in current genetic engineering. It can be used for applications such as improving crops (Jia and Wang, 2014), and it has a potential to cure HIV by removing HIV genetic material that has integrated into the host genome in human cells (Hu et al., 2014).
A number of automatic tools offer ways to identify CRISPR and analyze spacer diversity. Most tools search regularly spaced repeats in assembled genomes, for example: PILER-CR (Edgar, 2007), CRT (Bland et al., 2007), and CRISPRFinder (Grissa et al., 2007b). However, de novo assembly can be quite complicated, especially for repetitive regions. Crass is the first tool that can reconstruct CRISPR loci from raw sequence data (Skennerton et al., 2013). Crass requires that some of the reads contain two copies of a repeat. Our work aims at extending the work done in Crass, and it focuses on detecting CRISPR repeats from reads that are too short to contain two copies of a repeat.
The input to our algorithm is a raw sequence data of short reads, which we denote by
Following this observation, we analyze every frequent k-mer by constructing a partial overlap graph (Myers, 1995), using a hash-based method for overlap detection. These partial subgraphs are analyzed in order to determine whether they contain a long enough path, which corresponds to a long chain of overlapping reads. All the k-mers, which are deemed to be part of the same direct repeat, are subsequently clustered, and a consensus sequence is derived for every repeat.
We extended the approach of detecting CRISPR repeats directly from reads, which was introduced in Crass, and improved upon it by enabling a direct analysis of short reads. Compared to approaches that rely on assembled genomes, our algorithm can detect repeats in cases where assemblers produce very short contigs, due to multiple branches in the graph representation of the data set. Our work is relevant in practice, since many prokaryotic genomes were either sequenced with short reads, or have long CRISPR repeats.
2. Methods
2.1. Algorithm overview
The algorithm for detecting CRISPR repeats is composed of four main parts: (i) identifying frequent k-mers, (ii) analysis of frequent k-mers, (iii) clustering k-mers that are part of CRISPR repeats, and (iv) derivation of consensus sequences for the various repeats. We now describe these steps, as well as the underlying observation and the method used for indexing the reads.
2.2. Identifying frequent k-mers
The algorithm starts by identifying frequent k-mers in the genome. The length of k should not exceed the minimum known length of a repeat (24 bases), and could potentially be lower. Analyzing short k-mers is helpful, since some copies of the repeats are truncated or degraded. Furthermore, some of the reads contain only partial copies of the repeat, or a small number of errors.
We search for the maximal threshold t, such that every k-mer u, which is part of a CRISPR repeat, appears in the data set at least t times, with probability at least 1 − δ. In order to compute the threshold, we bound the probability of under-counting such a k-mer.
Let G be a (circular) genome of length |G|. Let u be a k-mer that is part of a CRISPR repeat. Let d be the minimum number of repeats in a CRISPR locus, ℓ the length of a read, c the coverage of the sequence data (the average number of reads covering every base), and n the number of reads. Suppose also that u appears in exactly d different copies of a repeat.
For each read i, 1 ≤ i ≤ n, we define the 0-1 random variable χi as follows:
Using the above notations, and the definition of coverage, we get that
Assuming reads are sampled independently and uniformly over the genome, all the χi variables are i.i.d 0-1 Bernoulli random variables. By definition, their sum is a lower bound for the number of occurrences of u in the data set. Therefore, we can find the value of the threshold by finding the maximum value of t that satisfies Prob(Sumn < t) ≤ δ. Since n is very large, we can use the normal approximation for the sum of independent Bernoulli random variables, namely
Using properties of normal distribution, we derive the following bound:
where Zδ satisfies Prob(Z ≤ Zδ) = δ, for a random variable
Due to the double-stranded nature of the genome, we count every k-mer together with its reverse complement. Note that with high probability, the counts of other repeating k-mers that are not associated with CRISPR repeats will also exceed the threshold.
Having a list of frequent k-mers, we can discard a great portion of
2.3. Analysis of frequent k-mers: basic observation
The next phase of the algorithm aims to determine, for every frequent k-mer, whether it truly belongs to a direct repeat of some CRISPR array. The analysis is conducted separately for every pair of a k-mer and its reverse complement. This phase is the heart of the algorithm. It is based on an observation that enables us to differentiate between CRISPR-related k-mers and other types of frequent k-mers. We start by defining a few required terms.
The term spacer edge comes from the fact that if reads ri and rj belong to a CRISPR locus, and assuming that they both contain two different consecutive instances of u, then the edge sequence contains an entire spacer (Fig. 2).
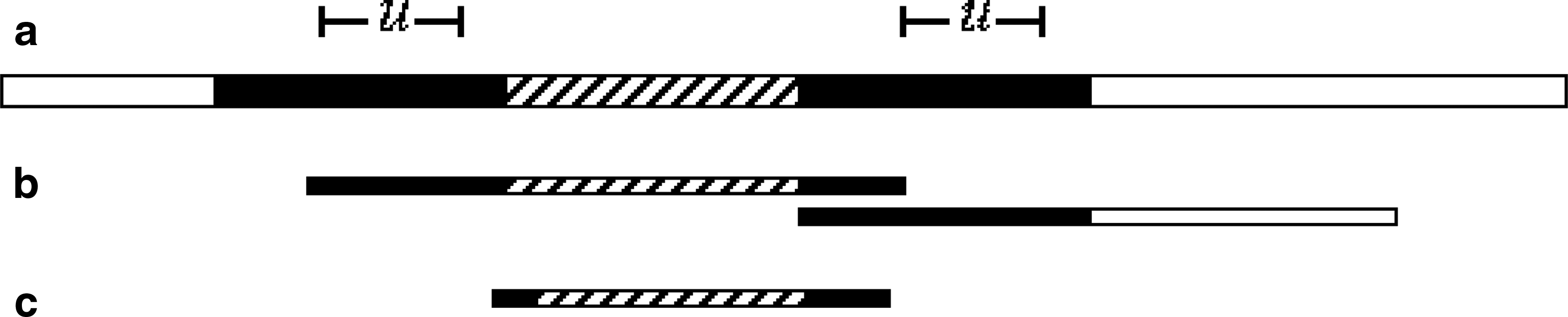
A spacer edge and its sequence:
The first part of the observation follows directly from the definition of the overlap graph, assuming high enough coverage. On the other hand, a frequent k-mer, which appears in a nonclustered and regularly interspaced repeat, does not typically form long paths in the induced subgraph, as the k-mer appears in distinct regions of the genome. In the case of a tandem repeat, even if the induced subgraph contains a long path, we expect the sequences of the spacer edges to be very similar. Low coverage and sequencing errors can influence whether long enough paths are seen in the induced subgraph. Hence, observation 1 is stated using probabilistic terms (see also Fig. 3).
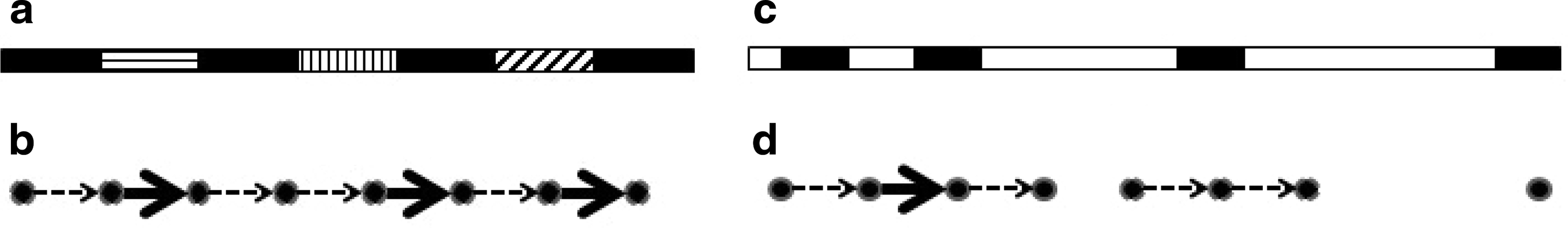
Illustration of observation 1:
2.4. Data indexing
The algorithm preprocesses the reads and constructs an index that enables efficient detection of overlaps that are greater than some threshold τ. Using a hash function F, the algorithm stores the hash values of all sufficiently long prefixes of reads in
2.5. Partial construction of overlap graph
Observation 1 enables us to analyze only parts of the overlap graph, since it requires nothing more than the evidence to an appropriate path. The algorithm does so by an interleaved process that both detects spacer edges and finds evidences for adjacency of pairs of spacer edges. When sufficiently many consecutive spacer edges are found, the k-mer is considered as part of a CRISPR repeat. This happens even though the path was not completely constructed.
These two procedures are performed in an interleaved manner. Figure 4 summarizes this process. Requirements on the expected length of the spacers can be enforced once the spacer edges are processed.
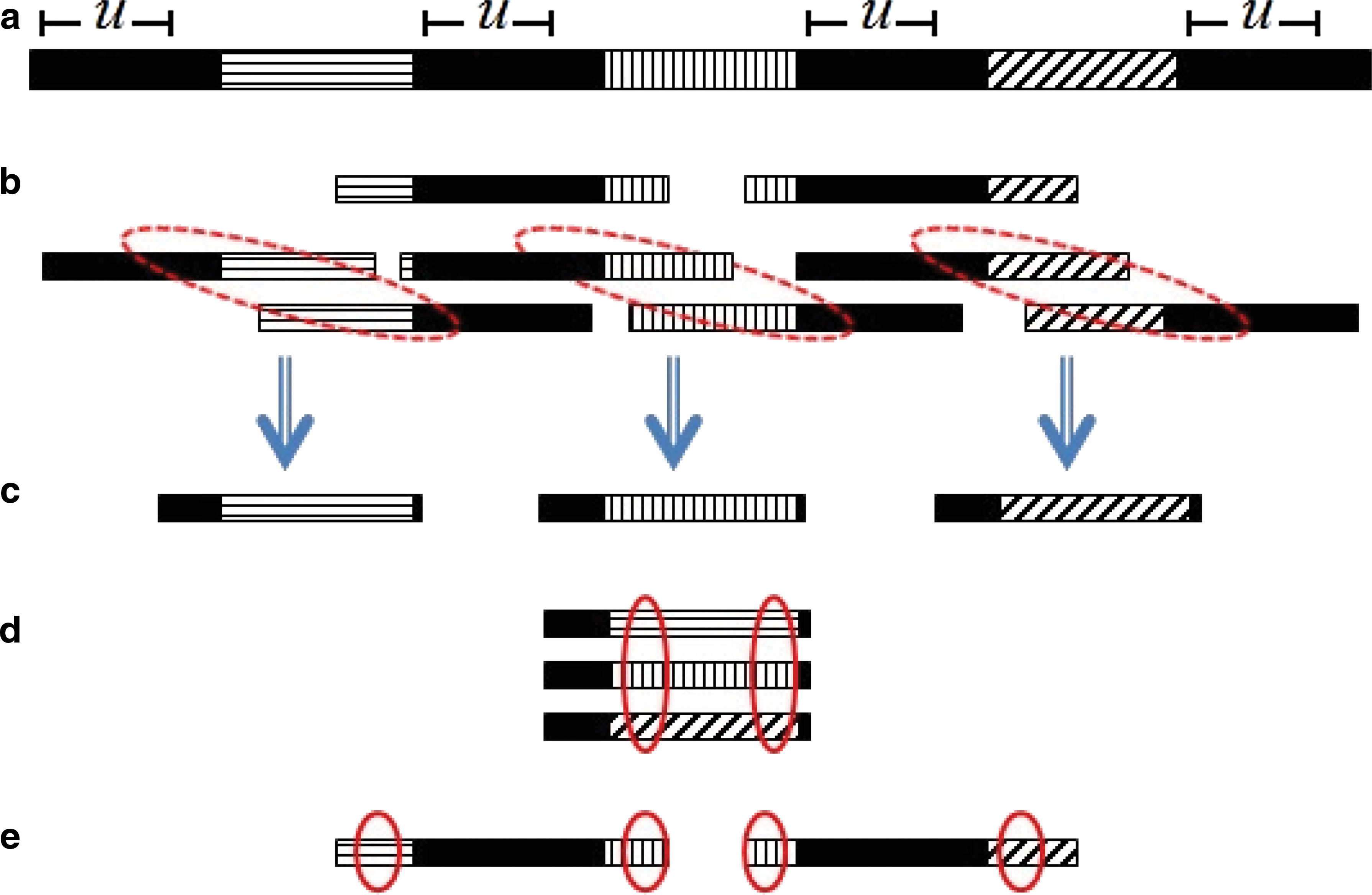
Partial construction of an induced overlap subgraph:
Detecting spacer edges and looking for adjacent pairs of them is a time-consuming task, as it might involve examining a substantial amount of overlaps. The algorithm can employ several optimization strategies if we assume that k-mers appearing in either a repeat or a spacer are not frequent in the rest of the genome. While this assumption might be violated in rare cases, it was empirically verified on the data sets used for assessing the algorithm.
Before describing the optimizations, we introduce a few terms. An isolated copy of a k-mer is an occurrence of the k-mer in the genome, which has no proximity to other copies of that particular k-mer. An isolated read is a read that contains an isolated copy of the k-mer being analyzed. By definition, an isolated read is not part of any overlap that is represented by a spacer edge. Nodes that represent isolated reads in the graph are referred to as isolated nodes.
Under the assumption mentioned above, we can impose an upper bound on both the number of isolated reads that are analyzed for every k-mer, and on the number of overlaps that are analyzed for every read (see Supplementary Method S1). In addition, overlaps are considered only for reads that do not contain the k-mer toward the end of the read; this optimization aims to focus on nodes with a higher probability of being adjacent to spacer edges. These optimizations have a substantial effect on the running time of the algorithm, as visualized in Figure 5.
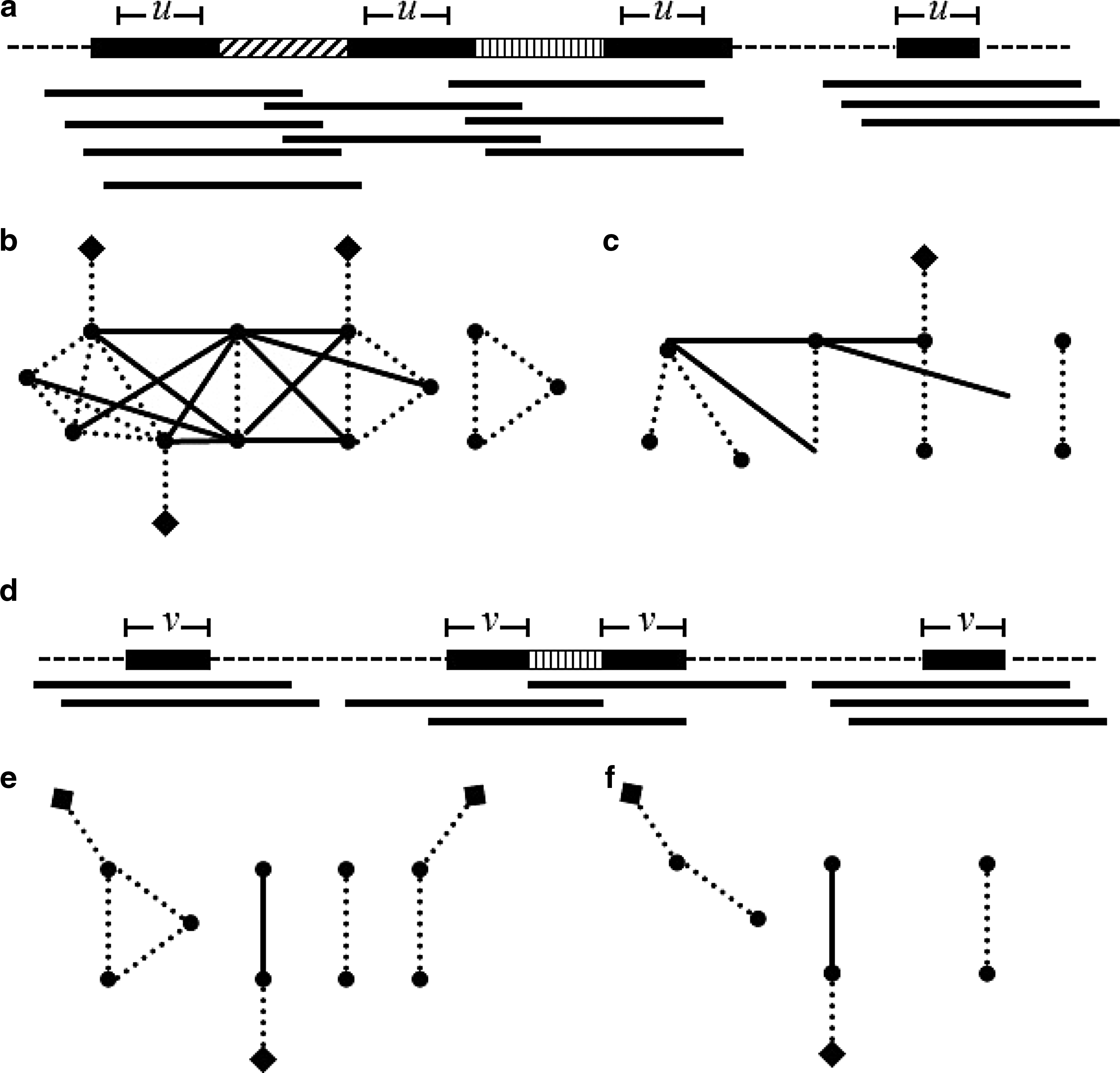
Partial overlap graph construction by sampling:
The partial construction of the overlap graph can be summarized as sampling relevant nodes and edges from the overlap graph. The k-mer is deemed part of a repeat if the sample induces a subpath with enough adjacent spacer edges. A pseudo-code of this process is described in Supplementary Algorithm S2.
2.6. k-Mer clustering
The collection of all frequent k-mers, which were found to be CRISPR related, typically contains several k-mers from each repeat. Before deriving the consensus sequence of each repeat, it is desirable to first cluster all k-mers of the same direct repeat together. While the clustering of k-mers can be done using a similarity test between the k-mers themselves, missing k-mers could potentially split a group into two separate clusters. Therefore, instead of using the k-mers directly, we employ a different approach, which uses the reads as templates.
An iterative process joins k-mers (or clusters of k-mers) that share a significant number of reads. The minimum number of common reads between two mergeable clusters is roughly the number of reads that are expected to cover the repeats of a CRISPR locus with a minimum number of repeats. This way, the problem of missing k-mers is circumvented by the presence of reads that contain several k-mers of the same direct repeat (Fig. 6).
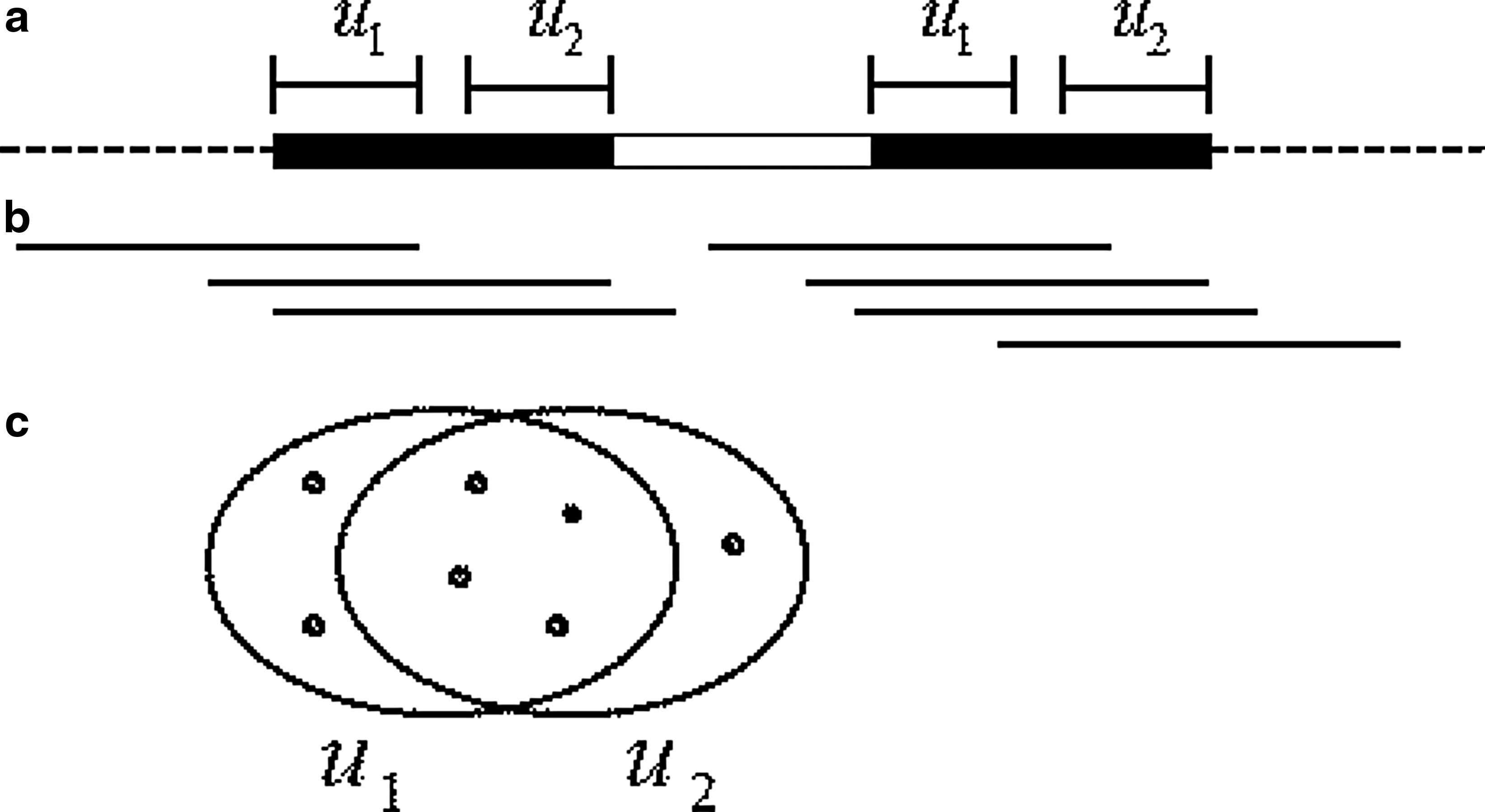
Clustering of two nonoverlapping k-mers, which belong to the same direct repeat, without knowing that the k-mers in between are also part of the repeat:
2.7. Repeat consensus derivation
In this phase, we use an alignment method, similar to the one used in Crass (Skennerton et al., 2013), for deriving the repeat consensus for every cluster. Ideally, every cluster should contain data related to a single CRISPR locus. However, since several CRISPR loci can share similar subsequences, clusters may contain two (or more) distinct, yet highly similar, direct repeats.
Before aligning all reads in a cluster, we employ a procedure that orients the reads. This is needed since the clusters contain reads from both strands. The orientation procedure iteratively orients k-mers and the reads containing them, until all k-mers and reads are consistently oriented. Reads that cause a contradiction are discarded (see also Supplementary Algorithm S3).
After orienting the reads in a cluster, it is possible to derive the consensus sequence of the direct repeat. Since conducting a multiple alignment of all reads in a cluster is computationally expensive, we choose one read as a seed and perform all alignments with respect to it. The read that is chosen as a seed is one that contains the largest number of frequent k-mers in the cluster. We align every read to the seed by finding the first k-mer that appears in both the read and the seed (in rare cases there are no common k-mers and the read is ignored).
After the completion of the alignment, the consensus sequence of the repeat is derived. For every position, we check whether there is a dominant base in all the aligned reads. The longest subsequence of positions with dominant bases is taken as the sequence of the direct repeat. In case of two dominant bases in one position, we derive more than one repeat sequence for this cluster. Direct repeats that violate the constraints on the length of the repeat can be discarded now.
The consensus derivation stage is expected to produce a correct sequence of a CRISPR repeat, even in cases where not all relevant k-mers were identified as CRISPR related. In such cases, most of the reads containing the repeat are still expected to be clustered together, so there is enough information to enable a correct derivation of the consensus sequence.
3. Results
As a proof of concept, we implemented a preliminary version of the algorithm in Python [apart from the k-mer counting phase, for which we used the Turtle program, written in C (Roy et al., 2014)]. We ran the code on several bacterial and archaeal data sets, with both simulated and real reads. We compared the outputs of the algorithm with the CRISPR repeats reported in the CRISPRdb (Grissa et al., 2007a). A comparison between the results of our tool and those of other methods was also conducted. In addition, we applied the algorithm to a metagenomic data set.
3.1. Simulated reads data
Table 1 shows the result of running our algorithm on simulated reads, derived from 20 bacterial and archeal genomes. For each genome, a data set of 76 base-long-reads was generated uniformly at random, with 20× coverage. The algorithm parameters were set as follows: k was set to 23; d, the minimum number of repeats in a CRISPR, was set to 3; and the probability of missing a repeat-related k-mer was set to 0.05. Consequently, the threshold for identifying frequent k-mers was set to 32 (Eq. 4). We used the value 50 as an upper bound on the number of isolated nodes that are sampled per frequent k-mer, and 200 as an upper bound on the number of edges sampled for each node (these values were set empirically). The minimum overlap required was 31 bases. A complete list of user-defined and hard-coded parameters can be found in the documentation of the code. We note that the output of our detection tool is a list of distinct direct repeats. The choice of read length was not arbitrary: Crass can process reads that are at least 76 bases. Our algorithm only requires that two overlapping reads would span a sequence containing two repeats and one spacer. This suggests that reads should be long enough to contain one repeat, plus half of the sequence of a spacer. For most genomes, the minimum read length requirement is therefore between 50 to 60 bases, and 70 bases for genomes with the longest repeat sequences known.
Precision and sensitivity rates are computed with respect to the number of distinct repeats only. Additional dominant variants of a repeat are not considered as false positives.
Boldface emphasizes the two most important colors.
The table details the results as well as the precision and sensitivity (or recall) of our tool. Note that while the numbers of repeats shown in the table are the actual numbers of CRISPR arrays in the various genomes, the precision and sensitivity rates are calculated using the number of distinct repeats.
Our algorithm achieves very good empirical results. It features perfect precision and did not report a single false repeat. Even though the tool analyzed thousands of frequent k-mers for each genome, it managed to exclude all irrelevant ones, including tandem repeats. In addition, our tool managed to identify the exact sequences of the repeats, including the exact boundaries, in all reported repeats except one (where an additional base, which is common to most spacers, was mistakenly added to the repeat sequence). In some cases, common variants of the consensus repeat sequence were also reported. Since these CRISPR repeats do appear in the genome, they are not counted as false negatives.
The algorithm also achieves a high sensitivity rate (88% in total). It managed to detect nontrivial repeats (such as repeats from short CRISPR arrays and repeats with multiple variants). Only a few repeats were missed. Some belong to very short CRISPR arrays, where mutations occur in most of the copies of the repeat, and some are almost identical to repeats from a longer array (causing two repeats with different abundances to be analyzed in the same cluster).
We compared the results of our tool to those produced by two other methods: the “assembly first” approach and Crass. Restrictions on the length of the repeats and spacers were set to high numbers, as in our tool. Crass (version 0.3.8) was not able to detect even a single repeat. The second method, which is currently the only way to detect CRISPR repeats from short reads, is to first assemble the reads and produce contigs, and subsequently apply a genome-based CRISPR detection tool. We ran Velvet (version 1.0.19) (Zerbino and Birney, 2008) to assemble the reads, and then CRT (version 1.1) to detect CRISPR repeats from the assembled contigs (both with their default settings, where the coverage of the data set was provided as input). Overall, our tool performed better than this method, both in the number of repeats detected and in identifying their boundaries. The assembly-first approach missed 3 repeats out of the 42 repeats that were detected by our tool. Some of the missed repeats belong to genomes with relatively long CRISPR repeats (36–40 bases long), which might add false branches to the de Bruijn graph produced by Velvet. Note that once Velvet outputs a contig that contains three (or more) copies of a repeat, CRT is usually able to detect the repeat. In addition to missing some of the repeats, a few false positive repeats were reported by Velvet and CRT. There was one case where the assembly-first method found a true repeat, which our tool did not find.
The running time and space consumption of the algorithm are listed in Supplementary Tables S1 and S2. The Python implementation is reasonably efficient: it takes a few minutes to run the code on a single instance, and between a few hundred MB and 2 GB of memory are used. A slight increase in the value of the frequent k-mer threshold improves the performance of the algorithm. An optimized implementation in a more efficient programming language is expected to reduce the run time.
3.2. Real reads data
We analyzed six real genomic sequencing data sets, downloaded from NCBI (SRR496816, SRR407315, SRR400622, ERR124723, ERR200050, and DRR018803), and one metagenomic data set (SRR638794). The length of the reads ranged between 71 to 101 bp, and we treated paired-end reads as two single reads. The frequent k-mer threshold was adjusted to the size of each data set. The results on the genomic data sets were similar to the ones obtained on simulated data: 100% precision and 90% sensitivity. One 50-base-long repeat was found by our tool, but not by the assembly-first approach. Crass detected only a single repeat (in one of the data sets with 100-base-long reads). As for the metagenomic data set (with 100 base-long reads), we were able to identify the same repeat that was identified by Velvet and CRT, while Crass did not find any repeats. This proves the potential of our approach for metagenomic data sets, in which assembly could be more challenging (Skennerton et al., 2013).
4. Conclusions and Future Work
This manuscript describes a novel approach for detecting CRISPR repeats from data sets of short reads. Prior to this work, identification of CRISPR repeats could be done either by analyzing assembled genomes, or by a read-level analysis using Crass, which requires that reads are not too short. Our work extends the ability to identify CRISPR repeats from raw sequence data, even when the reads are short. To the best of our knowledge, our tool is the first one capable of finding CRISPR repeats directly from such sequence data.
The algorithm exploits a property of overlap graphs that can distinguish reads originating from CRISPR loci from those that are not. Filtering and sampling are applied to detect k-mers that appear in CRISPR repeats, followed by clustering and consensus derivation processes, carried out in order to obtain the final sequences of the repeats. The algorithm incorporates hash functions to check overlaps efficiently.
A preliminary Python implementation yields excellent precision and good recall. A comparison of the results of our tool to the ones obtained by an assembly-first approach confirms the importance of our algorithm. This is due to the fact that assemblers might discard repetitive regions, or may not assemble them correctly. The graph representation of the reads (whether overlap graph or de Bruijn graph) might contain branches that prevent the assembly of long contigs. By way of contrast, our tool only searches for the existence of certain paths in the graph, and thus it is sometimes able to overcome ambiguities that general-purpose assemblers do not resolve.
Our approach is based on the tacit assumption of nearly uniform coverage and is thus applicable mostly to single species sequencing settings. However, metagenomic data sets often contain tens of millions of reads that were sampled with highly nonuniform coverage. Since the estimation of the threshold for frequent k-mers relies on uniform sampling, it can not be applied for such data sets. Better ways to assess the appropriate value of this threshold should be devised in such settings. For example, we have recently observed that analyzing the patterns in which frequent and nonfrequent k-mers coappear, may be useful for this purpose. Note that we cannot treat all k-mers as frequent, since then the downstream analysis will not be feasible.
There are several additional aspects of our work that may be improved upon: The overlap detection method could be made more robust to errors, even though errors are not as problematic here as they are with respect to de-novo assemblers. The consensus derivation method could also be improved, especially in cases where repeats with different abundances are clustered together. The ultimate goal of this line of research will seek a better assembly of complete CRISPR loci, not just the repeat sequence itself. Using the information gathered by this algorithm, we can analyze just a small subset of the data with algorithms that are targeted at repetitive regions.
We believe that techniques such as hashing and sampling can be further used in the domain of CRISPR detection. We described a specific implementation, but the concept presented in this article can be applied to assemblers operating in various frameworks. The sampling technique could also be replaced by other methods, such as incremental graph construction via random walks in the partial overlap graph. Code availability: see Ben-Bassat and Chor, 2016, for source code.
Footnotes
Acknowledgments
We wish to thank Rotem Sorek, Eran Halperin, Eran Mick, Rolf Backofen, and Omer Alkhnbashi for helpful suggestions. This study was supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.