
Abstract
Abstract
1. Introduction
P
Interactions between protein pairs could be modeled as a binary vector and therefore prediction of the interactions could be examined as a two-class classification problem. As a consequence, support vector machines (SVMs) were widely employed in PPI prediction. For examples, a SVM-based method using only the information of protein sequences was proposed in 2007 (Shen et al., 2007), a sequence-based method to predict yeast protein–protein interactions by combining auto covariance descriptor with SVM was proposed in 2008 (Guo et al., 2008), and a prediction method of protein–protein interactions from protein sequence using local descriptors was proposed in 2010 (Yang et al., 2010).
In PPI prediction, the performance of prediction might be affected by the selection of the positive and the negative datasets and lead to overestimated or underestimated results. Some properties had been investigated in the recent years. Ben-Hur and Noble (2006) showed that the distribution of testing examples reflects the performance of a predictor of PPI. However, they did not specify how the essential attributes of positive and negative dataset affect the performance of a predictor. Yu et al. (2010) found that the prediction result might be artificially inflated by a bias towards dominant samples in the positive datasets due to the presence of ‘hub’ proteins in the network. However, they did not note that the same result can occur for negative dataset.
In the PPI network, one protein might interact with multiple partners and lead to protein repetitiveness. Such object repetitiveness of the interaction dataset might cause SVM overfitting in high-dimensional omics data (Hall et al., 2009). As a consequence, it is necessary to study the effect of object repetitiveness of datasets on predicting results using the SVM-based method. In this study, we investigate the effect of protein repetitiveness on the performance of three SVM classifier methods, auto covariance (AC) (Guo et al., 2008), seven amino acids cluster (Sevenclus) (Shen et al., 2007), and local descriptor (Localdes) (Yu et al., 2010), both in the human PPI network and yeast PPI network. We found that removing repeated proteins in the positive dataset would decrease performance of prediction dramatically and performances of prediction increase with increasing protein repetitiveness (repeat number and repeat rate) of the negative dataset.
2. Methods
2.1. Protein pair dataset graph
A PPI network containing N proteins can be described as an undirected graph G = (V, E), where V = (1,…,N) is the set of N vertices representing N proteins, and E ⊆ V × V is the set of edges representing the protein pairs. A graph representing a protein pair dataset is called a protein pair dataset graph.
2.2. Power of the adjacency matrix and maximum matching of graph
A graph can be equally represented by a symmetric N × N adjacency matrix A = (ai,j) where the entry ai,j = aj,i = 1 if vertex i is adjacent to vertex j, otherwise ai,j = aj,i = 0. In graph theory, given a positive integer k, if A is the adjacency matrix of an undirected graph G, then the matrix Ak (i.e., the matrix product of k copies of A) has the property that the entry in row i and column j gives the number of undirected paths of length k from vertex i to vertex j in G. Accordingly, the entry in row i and column j of matrix W = A + A2 +…+ Ak, which is the sum of up to kth power of the adjacency matrix A, is the number of paths of length up to k from vertex i to vertex j in G, and the value can decide which vertices have contact level k with each other. Here, we take k = 6 according to the six degrees of separation of small-world theory. Let G = (V, E) be an undirected simple graph, a matching M = (V□, E′□) in G is a subgraph of G where no two edges in E′□⊆E share a common vertex of V. A maximum matching of G is a matching containing the largest possible number of edges of G.
In this article, the sum of up to 6th power of the adjacency matrix of a positive dataset graph is utilized to construct a series of negative datasets, and the maximum matching to construct the positive dataset without repeated proteins.
2.3. Protein repetitiveness: Repeat number and repeat rate of a protein pair dataset
To analyze the effect of protein repetitiveness of datasets on the prediction result, the concept of repeat number and repeat rate of a protein pair dataset is used. The occurrence frequency of a protein in a protein pair dataset minus one is called the repeat number of the protein, and the sum of all repeat numbers of proteins in the protein pair dataset is called the repeat number of a protein pair dataset. The repeat rate describes the average repeating numbers of every protein in the protein pair dataset. More precisely, let the degree sequence of a protein pair dataset graph with N proteins be d1, d2, …, dN, then s = d1 + d2 +…+ dN - N is the repeat number of the protein pair dataset and the quotient of s divided by N is the repeat rate of the protein pair dataset.
2.4. Positive datasets
The original PPI datasets are the core subset Hsapi20141001CR of H. sapiens, Scere20141001CR of S. cerevisiae, and sequence file fasta20131201 downloaded from the Oct 1, 2014 released updated interaction datasets of DIP (Salwinski et al., 2004). Proteins with sequences that could not be found in the sequence file or with sequence less than 50 standard amino acid residues were eliminated. Self-interaction was eliminated. Finally, the original positive dataset of H. sapiens, H, contained 4663 PPI pairs (3279 proteins) and the original positive dataset of S. cerevisiae, S, contained 4960 PPI pairs (2358 proteins) (Table 1).
The repeat removed datasets were created by the maximum matching algorithm which was implemented in the MatlabBGL package.
The negative datasets of correspondent positive datasets were made according to the six degrees of separation of small world theory.
2.5. Repeat protein removed positive datasets
As the original positive datasets of H. sapiens and S. cerevisiae contain repeating proteins, two repeat protein removed positive datasets, HM and SM, are constructed using maximum matching of the positive dataset graphs GH and GS. There are 1149 PPI pairs (2298 proteins) and 907 PPI pairs (1814 proteins) in the repeat protein removed positive dataset of H. sapiens and S.cerevisiae, respectively (Table 1). The maximum matching of a graph is implemented in the MatlabBGL package.
2.6. Negative datasets
Let PH be the protein set involved in the H. sapiens original dataset H, GH be the corresponding positive dataset graph of H, and the same is to PS and GS in the original S. cerevisiae positive dataset S. Clearly, the vertex set and edge set of GH are PH and H, and those of GS are PS and S, respectively. Let the adjacency matrix of GH be AH and that of GS be AS, the sum of up to 6th power of the adjacency matrices is calculated as follows:
By the property of adjacency matrix described above, if an entry in row i and column j of WH or WS is zero, then there are no paths of length up to 6 between vertices i and j in the corresponding positive dataset graph GH or GS, which means that the distance of vertices i and j is more than 6 in that graph. Due to the small-world property exhibited by biological networks, interacting proteins are expected to approach each other in the positive dataset graph. Conversely, true non-interacting protein pairs are more likely to correspond to the large shortest-path length (Ben-Hur and Noble, 2006; Trabuco et al., 2012). Therefore, according to the six degrees of separation of small world theory (Barabasi and Oltvai, 2004), if the entry in row i and column j of WH or WS is zero, the corresponding two proteins i and j, generally, do not interact.
Matrices WH and WS are used to construct series of negative datasets in the following way. Since there are 3279 and 2358 proteins in the original positive datasets H and S, respectively, the order of matrix WH is 3279 × 3279 and that of WS is 2358 × 2358. In order to assess effectively the prediction performance affected by datasets, considering the computational efficiency of SVMs, we randomly choose 200, 400, … , 3200 rows and the corresponding columns in WH, and 200, 350, … , 2300 rows and the corresponding columns in WS to construct two classes negative datasets corresponding to the original positive datasets of H. sapiens and S.cerevisiae, respectively. If the entry of WH or WS in a row and a column within chosen is zero, the corresponding protein pair is considered to be a candidate of non-interacting. In each case, by randomly choosing the same number of non-interacting pairs as the number of protein pairs in the corresponding original positive datasets, two original class negative datasets can be obtained and are highly incredible. The numbers of selected rows (columns) is set as the labels of the constructed original classes negative datasets (i.e., the original negative datasets of H. sapiens as NH200, NH400, …,and NH3200 and those of S. cerevisiae as NS200, NS350, …, and NS2300. The original positive dataset and the corresponding original negative datasets of H. sapiens are provided as Supplementary data S1 and that of S. cerevisiae is provided as Supplementary data S2 (supplementary material is available online at www.liebertpub.com/cmb).
The correspondent negative datasets of the repeat protein removed positive datasets were also created in the same way. Due to the size of network being smaller in the repeat protein removed positive datasets, we only randomly choose 200, 400, …, and 2200 protein pairs (XH200, XH400, …, XH2200) for H. sapiens (Supplementary data S3) and 200, 350, …, and 1700 protein pairs (XS200, XS350, …, XS1700) for S. cerevisiae (Supplementary data S4).
2.7. Protein–protein interaction prediction by support vector machine
Three typical feature representation methods, auto covariance (AC), seven amino acids cluster (Sevenclus), and local descriptor (Localdes), were used to construct the vector which represents protein. A protein pair is characterized by concatenating the vectors of two proteins, then the result vector is labeled with +1 for positive PPI and −1 for negative PPI. Predictors of three feature representation methods are constructed by the weka LibSVM package (the version weka-3-7-12jre) (Hall et al., 2009), respectively. Five cross-validation fold test options are used, and the regularization parameter C and the kernel width parameter g are manually chosen.
2.8. Measurements of prediction performances
The performances of all predication results were evaluated by true positive rate (TPR), false positive rate (FPR), precision, recall, F-measure, MCC, area under ROC (AUROC), and area under PRC (AUPRC), respectively. The relationships between the performance and protein repetitiveness under different datasets were evaluated by Pearson correlation and Spearman rank correlation.
3. Results
3.1. Repeat numbers and repeat rates of datasets
Table 2 shows the repeat numbers and the repeat rates of the original positive and repeat protein removed positive datasets of H. sapiens and S. cerevisiae. Due to the maximum matching algorithm, repeat number and repeat rate were zero in the repeat protein removed positive dataset. Negative datasets were created according to the positive datasets by the negative datasets construction algorithm and this might create repeated proteins. As a consequence, protein repetitiveness might be varied in different negative datasets and the relationships between SVM prediction performances and repetitiveness could be analyzed (Supplementary data S5 and S6).
3.2. Performance of prediction in the original datasets and repeat-protein removed datasets
In this study, we use TPR, FPR, precision, recall, F-measure, MCC, AUROC, and AUPRC to evaluate the performances of predictions of the original dataset and the repeat-protein removed dataset. By comparing performances of two datasets under different negative dataset settings, we found that TPR, FPR, precision, F-measure, MCC, AUROC, and AUPRC of the original dataset all outperformed those of the repeat-protein removed dataset both in H. sapiens and S. cerevisiae, respectively (Fig. 1 and Supplementary Figs. 1–5). In the AC model of H. sapiens PPI prediction, the performance of the repeat-protein removed dataset dramatically decreased from the negative dataset XH200 to XH800 and still hold low to XH2400 (Fig. 1, blue lines). On the other hand, in the original dataset, the performance gradually decreases with decrease of the repetitiveness of negative datasets (Fig. 1, red lines). This showed that removing repeat-protein in the positive dataset decreased the performance of prediction.
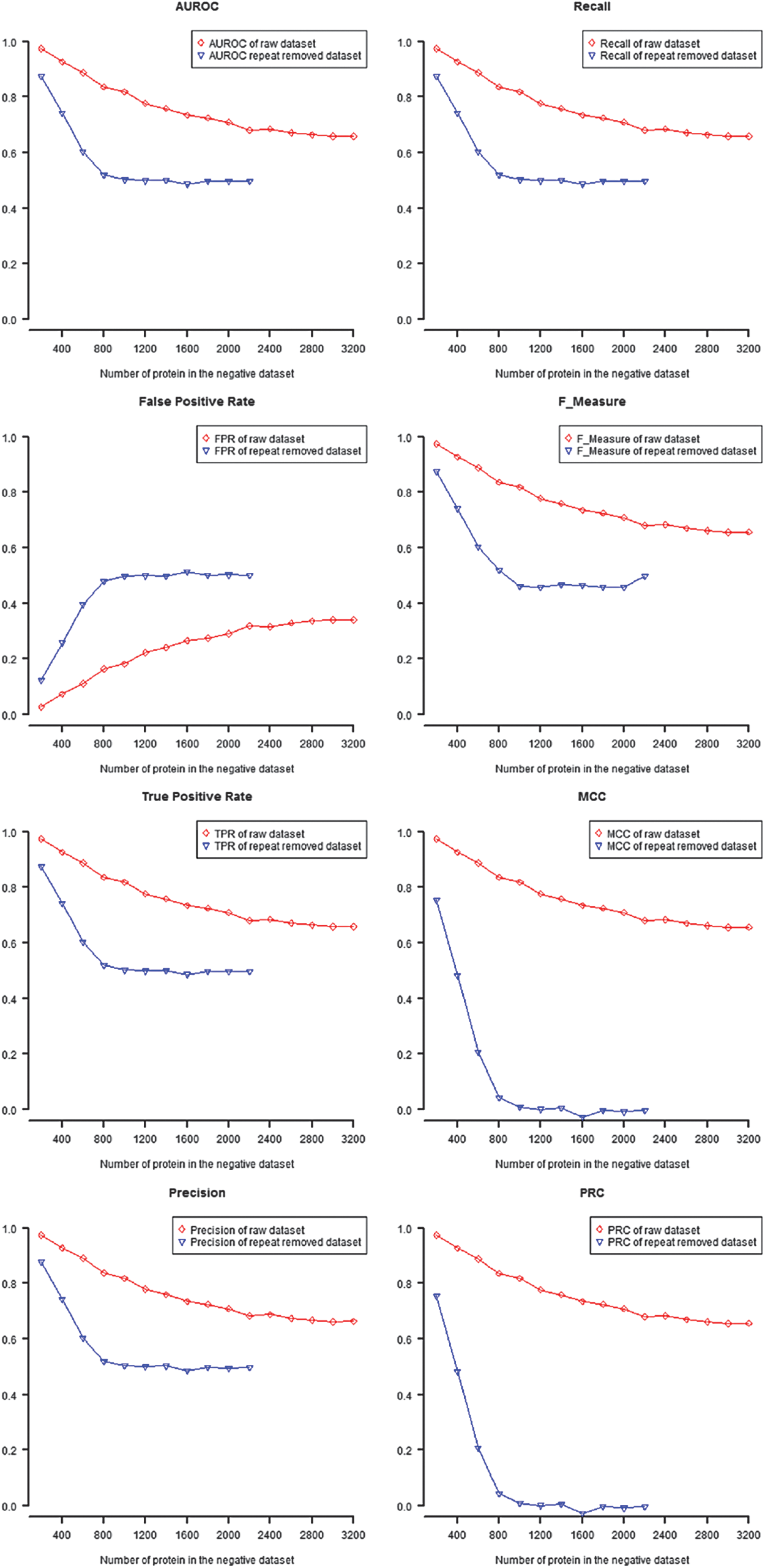
Performances of prediction in auto covariance model. TPR, FPR, precision, recall, F-measure, MCC, AUROC, and AUPRC were used to evaluate the performances of predictions of the original dataset and the repeat-protein removed dataset.
3.3. Performances of prediction decrease as protein repetitiveness and protein repeat rate decrease
Protein repetitiveness were varied in different negative datasets. The relationship between performances of prediction and protein repetitiveness of PPI datasets were evaluated by correlation analysis. Significant correlation between all performances of prediction and protein repetitiveness of the PPI dataset were shown (Table 3 and Supplemental Table). TPR, precision, recall, F-measure, MCC, AUROC, and AUPRC showed positive correlation and, on the other hand, FPR showed negative correlation in all models, species, and both in raw dataset and repeat-protein removed datasets (Tables 3, and 4, and Supplementary Tables 1–22). This showed that protein repetitiveness might affected performances of prediction no matter in the original positive dataset or in the repeat-protein removed positive dataset.
3.4. Distributions of repeated proteins in final datasets
In Table 5 and Table 6, we show parts of the degree distribution of raw dataset graphs and repeat-protein removed dataset graphs of H. sapiens, respectively. The maximum degrees of raw dataset graphs listed in Table 5 were from 180 to 200, while those of repeat-protein removed graphs in Table 6 were from 3 to 19. The low maximum degree of hub proteins in repeat-protein removed graphs showed that our repeat removing algorithm tended to remove hub proteins. The high maximum degree of hub proteins in raw dataset graphs lead to high repeat number and high repeat rate and this may contribute effect to high prediction performance in raw datasets (Table 5 and Fig. 1 red lines). In addition, although maximum degree of hub proteins were relatively low in repeat-protein removed dataset graphs, they also lead to high repeat number and high repeat rate due to large amounts of proteins with degree greater than 1 (Table 6). This implied that the presence of hub proteins or large amounts of proteins with relatively low degree in a dataset may both lead to in high prediction performance.
Discussion
In this study, the relationships between protein repetitiveness and performances of prediction of three SVM models were examined in H. sapiens and S.cerevisiae, respectively. We showed that removing repeated proteins in the positive or negative dataset would decrease performance of prediction dramatically, and performances of prediction increase with increasing protein repetitiveness of the negative dataset. Although we only investigated this relationship in SVM models with limited data, results might show a hint or a possibility that protein repetitiveness of the dataset is a bias factor of prediction performance. One possible reason for this phenomenon is the following: each vector in an encoding data of a dataset, other than label, is the concatenation of two vectors transformed from protein sequences of a protein pair. If repeat proteins in the dataset, they would create more duplicated vectors and the SVM predictor will learn from this repeated pattern as the same result. As a consequence, this might lead to high prediction performances of the SVM-based prediction methods because the positive and negative datasets might contain certain degree of repeat proteins.
In the previous studies, Yu et al. (2010) mentioned that the presence of ‘hub’ proteins that interact with many other proteins in the positive dataset leads to a strong bias that invalidates most performance estimates. We show that even removing those hub proteins in the positive dataset, such bias still contribute effects to performance due to protein repetitiveness of the negative dataset. Ben-Hur and Noble (2006) mentioned that restricting negative examples to non-colocalized protein pairs leads to a biased estimate of the accuracy of a predictor of protein–protein interactions and biased distribution of negative examples leads to over-optimistic estimates of classifier accuracy. In this study, we further show that the presence of hub proteins or large amounts of proteins with relatively low degree in a dataset may both lead to high prediction performance.
Although we only analyzed the relationship between protein repetitiveness and performances of PPI prediction, the results might be applied to other kinds of predictions such as gene–protein interactions, RNA–protein interactions, gene–gene interactions, gene–disease interactions, RNA–disease interactions, protein–DNA interactions, drug–protein interactions, and cell–cell interactions (Cheng et al., 2015; Koo et al., 2013; McKinney et al., 2006; Muppiral et al., 2011; Piro and Di Cunto, 2012; Rao et al., 2014; Westra et al., 2007; Xu et al., 2015; Zhang et al., 2014; Zou et al., 2013). The most similar characteristic between these predictions and PPI prediction was object repetitiveness. As a consequence, performances of SVM prediction might be also affected by repetitiveness in the positive dataset or negative dataset.
In this study, we only conducted SVM on PPI prediction and analyzed the effect of protein repetitiveness on performances. The mechanism of these effects on performances of SVM prediction is still not clear. Improvement or modification of the SVM algorithm to avoid the effect of repetitiveness on performances of prediction is still worthy of future investigation.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.