
Abstract
Abstract
In the two transmembrane protein types, outer membrane proteins (OMPs) perform diverse important biochemical functions, including substrate transport and passive nutrient uptake and intake. Hence their 3D structures are expected to reveal these functions. Because experimental structures are scarce, predicted 3D structures are more adapted to OMP research instead, and the inter-barrel residue contact is becoming one of the most remarkable features, improving prediction accuracy by describing the structural information of OMPs. To predict OMP structures accurately, we explored an OMP inter-barrel residue contact prediction method: OMPcontact. Multiple OMP-specific features were integrated in the method, including residue evolutionary covariation, topology-based transmembrane segment relative residue position, OMP lipid layer accessibility, and residue evolution conservation. These features describe the properties of a residue pair in different respects: sequential, structural, evolutionary, and biochemical. Within a 3-residues slide window, a Support Vector Machine (SVM) could accurately determinate the inter-barrel contact residue pair using above features. A 5-fold cross-valuation process was applied in testing the OMPcontact performance against a non-redundant OMP set with 75 samples inside. The tests compared four evolutionary covariation methods and screen analyzed the adaptive ones for inter-barrel contact prediction. The results showed our method not only efficiently realized the prediction, but also scored the possibility for residue pairs reliably. This is expected to improve OMP tertiary structure prediction. Therefore, OMPcontact will be helpful in compiling a structural census of outer membrane protein.
1. Introduction
O
OMPs constitute no more than 2% of all solved structures (Berman et al., 2000). However, it is reported that the number of recorded protein sequences are almost a hundred times larger than that of structures (Kajan et al., 2014), so that protein structure prediction provides a practical approach to push forward structure-based OMP research. Major advances have been made for soluble protein structure prediction, but it is still a challenging problem to deal with transmembrane proteins, especially the OMPs.
Homology modeling methods were tried in transmembrane protein tertiary structure prediction because they performed well with soluble proteins, such as SWISS-MODEL (Arnold et al., 2006), and succeeded in modeling human transmembrane protease 3 using remote homology templates, MEDELLER. Kelm et al. (2010) tried to improve the prediction accuracy by separately predicting the transmembrane cores and loops. Great achievements were derived for G-protein-coupled receptors (GPCRs) protein family (Becker et al., 2004; Kalani et al., 2004; Shacham et al., 2004; Trabanino et al., 2004; Zhang et al., 2006; Michino et al., 2010). However, the prediction accuracy of homology modeling depends on the number of homology proteins with the target protein, which has limited performance applied to OMPs. Fold recognition method is a better choice for OMP structure prediction theoretically; it has been widely applied to structure prediction for remote homology soluble proteins (Murzin and Bateman 2001; Ahmad et al., 2003; Cheng and Baldi 2006; Liu et al., 2007; Zhou and Skolnick 2010).
Our previous work with TMFR (Wang et al., 2013) first tried to use fold recognition methods for both OMP and α-helical transmembrane proteins. The OMP folds were more accurate for the structure prediction, but it depended highly on the predicted topology structure; structural features should be enhanced. In a recent report, a novel structure prediction method (Hayat et al., 2015) adopted OMP sequence evolutionary coupling to predict H-bonded residue pairs, and then constructed the all atom structures. The method elucidated that the evolution covariation of OMP sequence is helpful to discover the structural property from inter-barrel contact.
Inter-residue contact exists between a pair of residues in one protein, when they can strongly exhibit the protein's spatial constitution. OMPs have more regular structures than soluble proteins. The inter-barrel residue contacts could well explain the protein conformation, so that inter-residue contact prediction will obviously improve the prediction of protein structure. Many relevant efforts had been made first for soluble proteins and then for transmembrane proteins. Aiming for the final use, a residue contact prediction is expected to generate directly from the protein sequence itself. Many particular machine learning methods had been applied to the field, including neural networks (NN) based methods (Tegge et al., 2009; Walsh et al., 2009), support vector machine (SVM) based methods (Wu and Zhang 2008), and other novel methods (Li et al., 2011; Di Lena et al., 2012; Eickholt and Cheng 2012; Ekeberg et al., 2013).
These methods tried to find sequence patterns related to inter-residue contact, but have not yet satisfied the high accuracy requirement for protein structure prediction, according to Monastyrskyy et al. (2011); the accuracy of inter-residue contact prediction is in the range of 25%–35%. Recently, some research (Fuchs et al., 2009; Nugent et al., 2011; Nugent, 2015) predicted inter-residue contacts for α-helical transmembrane protein and improved the accuracy comparatively, but there is no available predictor for OMP inter-barrel contact.
A breakthrough happened when evolutionary information derived from multiple sequence alignment (MSA) was applied to the contact prediction. A series of methods explored this approach (Vicatos et al., 2005; Morcos et al., 2011; Cocco et al., 2013). PSICOV (Jones et al., 2012) and EVFOLD (Marks et al., 2011) are more accessible by offering stand-alone executable tools to calculate the evolutionary covariation. Notably, a recent work, FreeContact (Kajan et al., 2014), improved the performance of the two methods. Evolutionary covariation has been applied to transmembrane protein research, such as mining evolutionary hubs in GPCR families (Pele et al., 2014), or directly predicting all-atom 3D structure for OMPs (Hayat et al., 2015) using evolutionary covariation. Evolutionary covariation-based methods work efficiently only when a high quality MSA is available, which is a disadvantage for OMPs. Another approach, DNcon (Eickholt and Cheng, 2013), integrated multiple protein sequence-based features to predict inter-residue contact; it achieved a certain accuracy without relying on the MSA itself.
We then developed an inter-barrel residue contact predictor OMPcontact to further improve OMP structure prediction and structure relevant research. Multiple OMP-specific features had been integrated in the method to describe the properties of OMP comprehensively, where evolutionary covariation and topology are structural features; residue evolution conservation and residue relevant position describe the sequence pattern, while lipid layer accessibility reflects the biochemical property of OMP. These features restrain the prediction to the right way, and avoid the disadvantages caused by the shortage of OMP samples. Adaptive evolutionary covariation methods were selected to implement the prediction, and a SVM (Support Vector Machine) model were used to test the performance of OMPcontact against our dataset.
2. Materials and Methods
2.1. Dataset
PDBTM (Protein Data Bank of Transmembrane Proteins) (Tusnady et al., 2005) can determine the topology structures for all the transmembrane proteins using an automated algorithm (TMDET) (Tusnady et al., 2004), and update the data weekly following the PDB (Protein Data Bank). It is the most comprehensive current transmembrane protein structure data source. There are 317 OMP entries at the time of study, among them, 82 entries are non-redundant. By removing the entries that have incomplete tertiary structures, and those redundant chains in each entry, a 75 OMP sequence dataset was selected for testing, see in Supplementary Table S1 (Supplementary material is available online at www.liebertpub.com/crb).
2.2. Topology structure feature
As a crucial key to discovering the structure of OMP, topology structure can better describe the specific structure different in OMPs from other soluble proteins. Those transmembrane segments have significant biochemical properties since they are located in particular parts of biomembrane, and the number of them is a strong signal to indicate the protein family to which the OMP belongs. Inter-barrel contacts exist between the residues on adjacent transmembrane segments to construct a stable beta-barrel, as shown in Figure 1. Inter-barrel residue contacts are well-distributed on transmembrane segments, most of them formed by one or two hydrogen bonds. Obviously, topology structure of OMP will be theoretically the most helpful to find the inter-barrel contact.
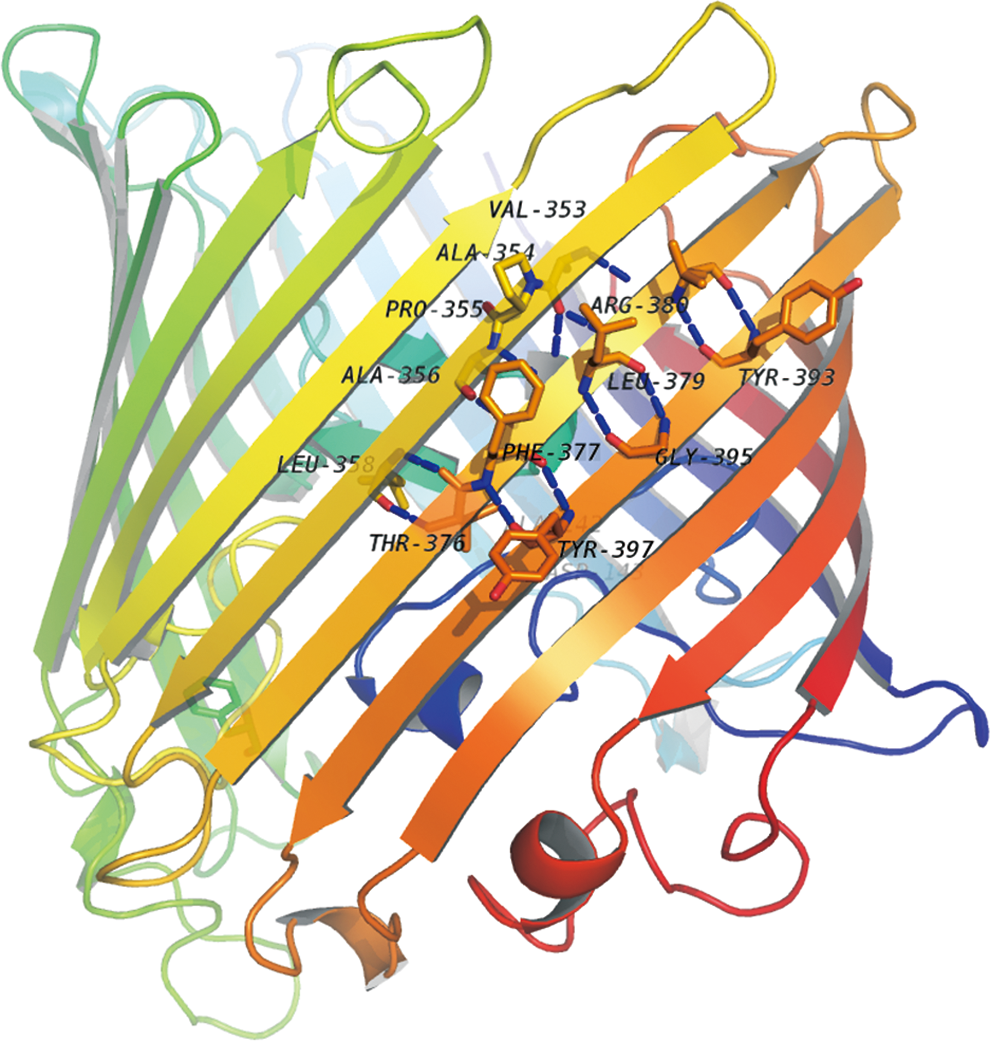
An example of native inter-barrel contact in OMP. The inter-barrel residue contacts bond the transmembrane segments to construct a stable beta-barrel of a OMP. A blue dash line in the figure represents an inter-barrel contact. There are two or more contacts between any two transmembrane segments.
The native topology structures can be accessed from PDBTM. It was used to train the contact prediction models, and filter the inter-barrel contacts from other contacts with at least one residue not in any transmembrane segment. Predicted topology was used to predict the inter-barrel contact. There are several OMP topology prediction methods available (Singh et al., 2011; Hayat and Elofsson 2012). Considering the prediction performance and the computation dependency, BETAWARE (Savojardo et al., 2013) was chosen in our work since it achieves a high prediction accuracy, and offers a stand-alone program for prediction. Both the native and predicted topology structures were unified to the same format, in which the residues in transmembrane segments were identified using character “T”, those residues outside the outer membrane used character “o”, residues inside the outer membrane used “i”, and the others assigned to “U”. Then, all the characters composed a sequence equal to the original protein sequence in length.
2.3. Residue relative position feature
Adjacent transmembrane segments are anti-parallel to cross the outer membrane, so that the inter-barrel contact will appear highly relevant to the position of the residues existing in the transmembrane segments. As shown in Figure 1, the residue ARG in position 380 near the N-terminal side of corresponding transmembrane segment is likely to contact the residues closing the C-terminal of the following transmembrane segment, residue TYR in position 393, which is in that area to contact with it.
For each residue pair to be predicted, the relevant position of the residue near the N-terminal side was counted from the N-terminal end of the corresponding transmembrane segment, and the other residue was counted from the C-terminal side when it is in the adjacent transmembrane segment, otherwise, the relative positions were assigned to −1 for both residues. According to the definition, two residues are more possible to have contact only when they are in the adjacent transmembrane segments, and the relative positions are close to each other. Relative position is a topology-based feature.
2.4. Evolution conservation feature
Multiple alignments against the large-scale protein sequence database could indicate residue evolution conservation for most sequence, which have plentiful homology proteins. For OMPs, there are less homology members, but almost all OMPs are homologous to each other (Arnold et al., 2007); multiple alignment could still generate acceptable results. PSI-BLAST (Altschul et al., 1997) were used here to generate the sequence-based profile against NCBI's non-redundant sequence database (NR) by three iterations. The evolutionary conservation of a sequence was present in a PSSM (position specific scoring matrix) profile PM, in which an element PM[i,j] represents the log-odd frequency that residue j happens in the sequence position i.
2.5. Evolutionary covariation feature
The conserved relationship between a pair of residues could be described as evolutionary covariation, which more directly reflects the inter-barrel contract, since most inter-barrel contacts involved residues are covariate. Four evolutionary covariation methods were chosen to calculate the covariation for all the residue pairs of each OMP in the dataset, despite whether they are in the transmembrane segment or not. Among them, ELSC, MI, OMES methods generated the evolutionary covariation using the Java code provided by Fodor at http://www.afodor.net, and EVFOLD results were generated using Python version of FreeContact (Kajan et al., 2014) for its performance and convenient. Covariations calculated for different protein sequences are not in the same value range; a standardization using z-score makes them comparable with each other, so that all the covariation could be input to the contact prediction as a valuate feature.
2.6. Lipid layer accessibility feature
Compared to soluble proteins, transmembrane segments of OMPs face the lipid layer of outer membrane. Hydrophobic residues are rich in the outside surface of beta-barrel against such environment, which significantly reflects the property of transmembrane distinguished with the sequence-based features. Therefore, accessible surface area (ASA) is much different for OMPs to be predicted. We used a recent published method MPRAP (Phatak et al., 2011) to generate the ASA for all the residues in each sequence in our dataset. The predicted ASA were directly used in contact prediction as an element in the input feature vector.
2.7. Support vector machine
The multiple features selected formed a non-linear dependency feature vector. The SVM method (Noble, 2006) is a promising classifier to determinate the residues that have contact. In addition, the prediction result of SVM could be directly input to the OMP structure prediction as a valued feature of contact.
A 3-residues slid window was used to compose the feature vector for each residue, for the contact prediction between residues Ai and Bj, the features abstracted from residues (Ai−1AiAi+1) and (Bj−1BjBj+1), where, i,j is the sequence position, and i ≠ j. According to the slid window, a SVM input Vi,j is given as following:
where (T, R, E, S) i is the topology, relative position, evolution conservation, and lipid layer accessibility features of residue in position i, and Ci,j is the predicted evolutionary covariation between residue i and j. The size of slid window is set according to the properties of OMP. When the size is greater than three residues, there would be more false positive predictions produced, because many non-transmembrane loops are shorter than three residues, and neighbor inter-barrel contacts are generally very close. A bigger slid window would have too many overlaps among the contact residues.
A Python package in LIBSVM (Chang and Lin, 2011) toolkit version 3.2 was used to generalize the SVM model with radial basis function (RBF) kernel; the corresponding parameters (C, g) were optimized using the grid-search method against the training data separately in the cross validation process. Here, all the contact residue pairs in the transmembrane segments from the whole dataset were picked up as positive samples, and the similar number of non-contact residues in transmembrane segments were selected as negative samples. The positive samples and negative samples were randomly picked up with same number to compose five sample groups, which had been applied to a 5-fold cross validation for SVM prediction performance.
3. Results and Discussion
3.1. Evolutionary covariations partly matches the native contact
As an important feature to classify the inter-barrel contact residue pair, evolutionary covariation is expected to match the native contact perfectly to achieve higher accuracy. Meanwhile, choosing a proper evolutionary covariation method is crucial for the prediction. For that purpose, we calculated the evolutionary covariation for all the residue pairs in dataset using four different methods, ELSC, EVFOLD, MI, and OMES. For each method, the calculated evolutionary covariations are not in the same value space for different protein sequence; z-score was used to standardize the values. A standardized evolutionary covariation with value greater than 4 was considered that the corresponding residue pair is possible to have a residue contact (Atchley et al., 2000). Meanwhile, native contacts of each protein in dataset were calculated according to the criterion that the atom distance between the heavy carbon of residues is less than 5.5 Å.
The distribution of evolutionary covariation and native contact are shown in Figure 2, where red points correspond to the residue pairs with higher evolutionary covariation, and the gray points correspond to those pairs having native contact. Notably, 4QD6, 3SYB, and 2IAH were OMP samples randomly selected whose residue sequence length was close to 200, 500, and 800, respectively. The OMPs in the same protein family have the same number of transmembrane segments, and their residue sequences are close to each other in length. Those OMPs having similar sequence length are likely in the same protein family. It is obvious that the three samples are not in the same protein family with each other, which would be better to reveal the property of each evolutionary covariation method comprehensively.
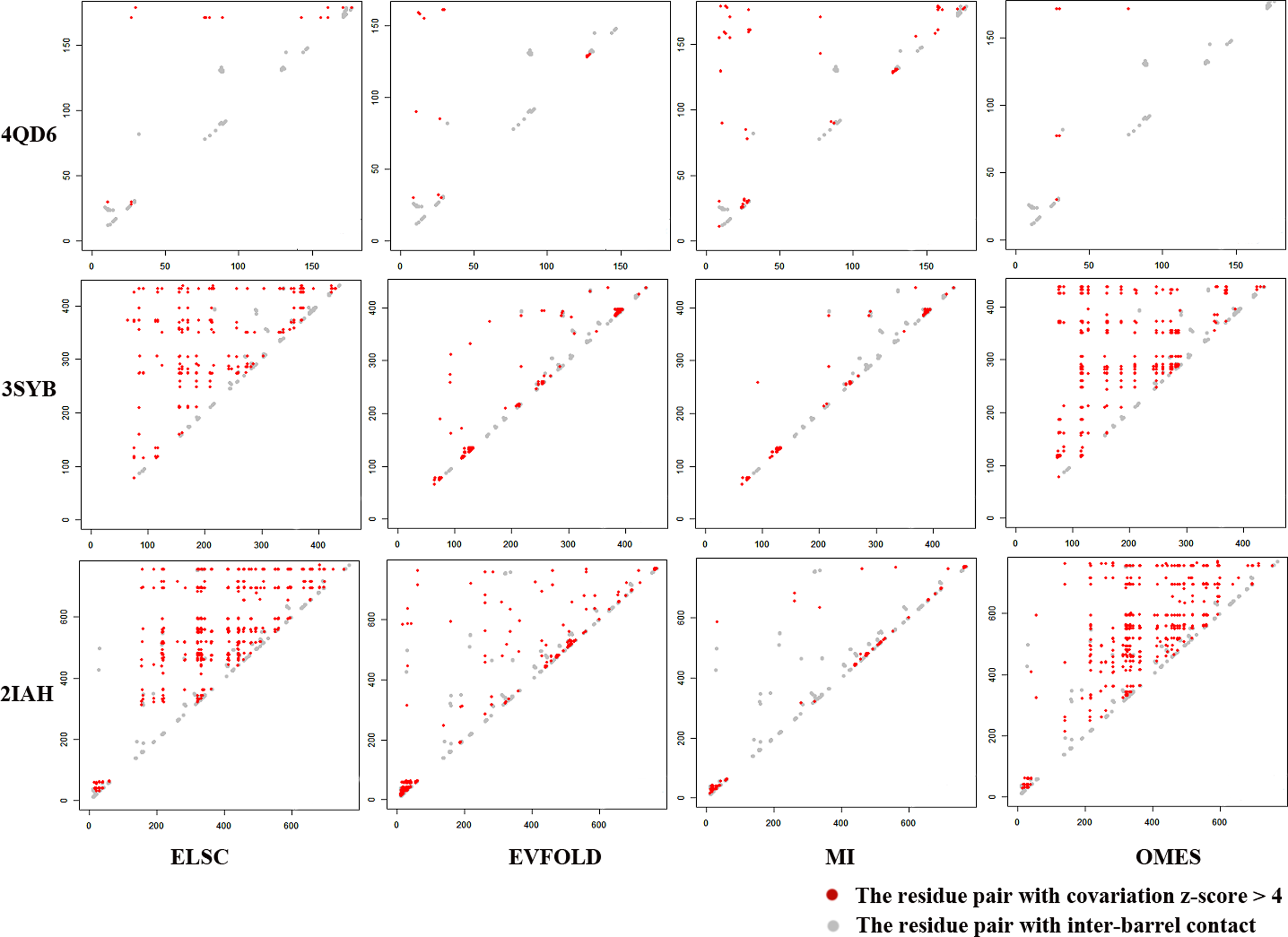
The distributions of standardized evolutionary covariation and native contact. Evolutionary covariation was observed relative to the length of residue sequence, OMP 4QD6, 3SYB, and 2LAH were randomly selected according to their sequence length of 200, 500, and 800 residues, respectively. The distributions of covariation residue pairs calculated by EVFOLD and MI were more similar to that of native inter-barrel contact, while the other two methods gave more irrelevant pairs. There was no method could perfectly match the native inter-barrel contact.
Obviously, ELSC and OMES were similar in the covariation distribution, where more residue pairs were considered to be covariate, which are range far more over the number of native pairs, a result because most OMPs have remote homology. There would have more sequence similarity in the multiple alignments that were used to generate the covariation. Neither method could distinguish whether those pairs of residues were evolutionarily covariate or contacting with each other. That is why many contact methods did not use the z-score threshold to classify the contact residues, but adopted the approach of ranking z-score. However, problems still existed when the target had a small sample dataset, such as OMPs. Comparatively, EVFOLD and MI generated the covariate residue pairs matching the native contact distribution much better, and had the similar number of native contact pairs.
It was found that no method could perfectly match the point distribution of native contact, even though there might have been an approach to make distributions better matched. It will not be denied that residue contact could not be found only depending on the evolutionary covariation itself, otherwise, protein structure prediction would be much easier to achieve higher accuracy. But evolutionary covariation is still a vital key to discovering residue contact as a feature, since they are highly relevant in biology. A pair of residues having contact can possibly covariate with each other, and those covariate residues are likely close to contact residues. Based on the above acknowledgement, we believe evolutionary covariation could be used to predict inter-barrel residue contact as a promising feature, and adopted a slid window to utilize it accordingly as mentioned.
3.2. Threshold of SVM models
Four SVM models had been applied to inter-barrel contact prediction using the four covariation methods. For each pair of residues in the target OMP sequence, a four SVM score would be given after calculation. These scores were possibile if the two residues might have contact predicted by according SVM model; they were in the range of [0, 1]. A threshold should be scientifically defined to determinate which pairs will have contact, that means if the SVM score is greater than the threshold, the corresponding residue pair is more probable to have a contact.
To derive reasonable thresholds for each SVM model, tests were made by changing the threshold from 0.3 to 0.74, and then comparing the prediction accuracy. These were described by the Matthews correlation coefficient (MCC), sensitivity (TPR), specificity (SPC), and accuracy (ACC). In Figure 3, the prediction accuracy changes of all SVM models were shown in polylines, MCC, TPR, SPC, and ACC are in different colors. It was found so consistent with Figure 2 that ELSC and OMES had the similar polylines, while that is similar between EVFOLD and MI.
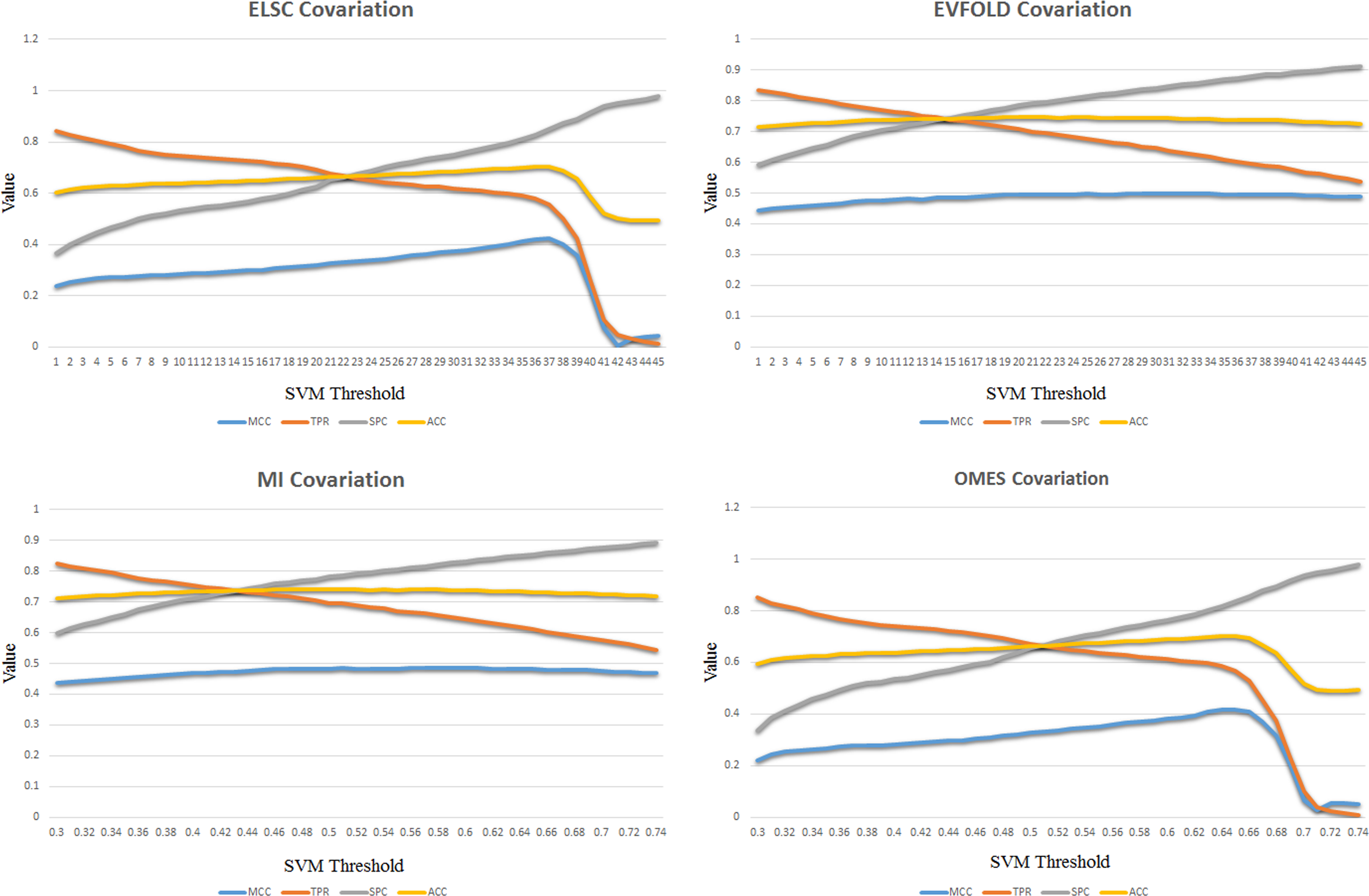
SVM performance using four evolutionary covariation methods. The SVM model predicted the inter-barrel contact with different performance according to the selected covariation methods. By changing the thresholds of SVM prediction result, step 0.2 in range of (0.3, 0.74), the performances of four evolutionary covariation methods are shown, respectively, where MCC (Matthews correlation coefficient), TPR (sensitivity), SPC (specificity), and ACC (accuracy) are used to describe the performance.
Using the ELSC or OMES, prediction accuracy declined when the thresholds were greater than a particular value, along with sensitivity raised. The phenomenon exhibited that more native contact pairs were excluded to their group when the standard became stricter, the covariations calculated by ELSC and OMES methods did not follow the order of the possibility to contact. On the contrary, EVFOLD and MI made more smooth polylines that the prediction accuracies were reasonable changed, the true positive ratio steadily decreased along with threshold increased, and the true negative ratio did contrarily. The test proved EVFOLD and MI covariations were more acceptable for inter-barrel residue contact prediction for OMPs.
OMPcontact was designed to offer the possibility of contacts to improve OMP structure prediction. Accuracy and stability are equally important for the purpose, while the ability to determine the contact dose directly, not comparing the vital demand. The use of EVFOLD or MI satisfied the requirements since the relativity between contact prediction score and contact possibility had been well reflected, as well as the prediction accuracy was stable on a high level. It means that the prediction result of OMPcontact could be used directly as a tertiary structure relevant feature for OMP structure prediction, such as sequence to structure alignment, fold recognition.
The inter-barrel contact prediction accuracies are listed in Table 1, the corresponding SVM thresholds are shown, along with accuracies for each evolutionary covariation method. Here, MCC value was considered to describe the prediction accuracy more comprehensively instead ACC value alone. Predictions using ELSC and OMES had the same ACC (0.72) and MCC (0.46) value using the same SVM threshold (0.65), which could be seen from the performances of two covariation methods to match native contact or determinate SVM threshold. They even share similar TPR and SPC in the same way. EVFOLD and MI derived equal prediction accuracy, but it was marked higher than the former, which could be seen from their MCC (0.52) and ACC (0.76) values. It should be noted that these two covariation methods had improved the prediction accuracy, while they used a lower SVM threshold (0.44). TPR was raised almost 20% compared to the other two covariation methods, and SPC dropped down nearly 10%. More true positive and false negative predictions had been made. EVFOLD and MI methods were more efficient to reflect the relationship between the residue contact and evolutionary covariation.
ACC, accuracy; MCC, Matthews correlation coefficient; SPC, specificity; TPR, sensitivity.
3.3. Prediction performance
To present the prediction performance intuitively, parts of the predicted inter-barrel residue contacts were marked on the tertiary structures of selected proteins: chain P of 1A0S, 3QRA, and 2LME, where the correctly predicted contacts were connected by blue dash lines, while red dash lines were used for incorrect ones, and purple dash lines were used for missing ones. They represented the true positive prediction, false negative prediction, and false positive prediction, respectively. Three sample proteins are representational OMPs in the dataset different with each other, and the details will be introduced in the following paragraphs.
Three same sequence chains in 1A0S separately compose a beta-barrel structure and make a compound. For chain P, there are 18 transmembrane segments in its sequence, namely the beta sheets. It is a relatively larger protein in OMPs, and also a typical one with regular tertiary structure. The contact prediction was made more accurately to this class of OMP, because their sequences are much more disciplinary with similar length of transmembrane segments, less structures in non-transmembrane segment, and the evolutionary covariation could be more clearly observed. For the same reason, corresponding topology prediction was more accurate, and the other features seemed to be better described. Therefore, higher predictions were made on these OMPs. As shown in Figure 4, all the native contacts were found in the sample part of 1A0S_P, while a few false negative predictions were made. These incorrectly prediction happened close to those contact residues, sometimes even overlapped, as residue ALA in sequence position 354. From 1A0S, we could conclude that these types of OMPs would derive higher prediction accuracy contributed by high TPR, and it might be further improved by cutting off a few positive predictions when they are too dense.
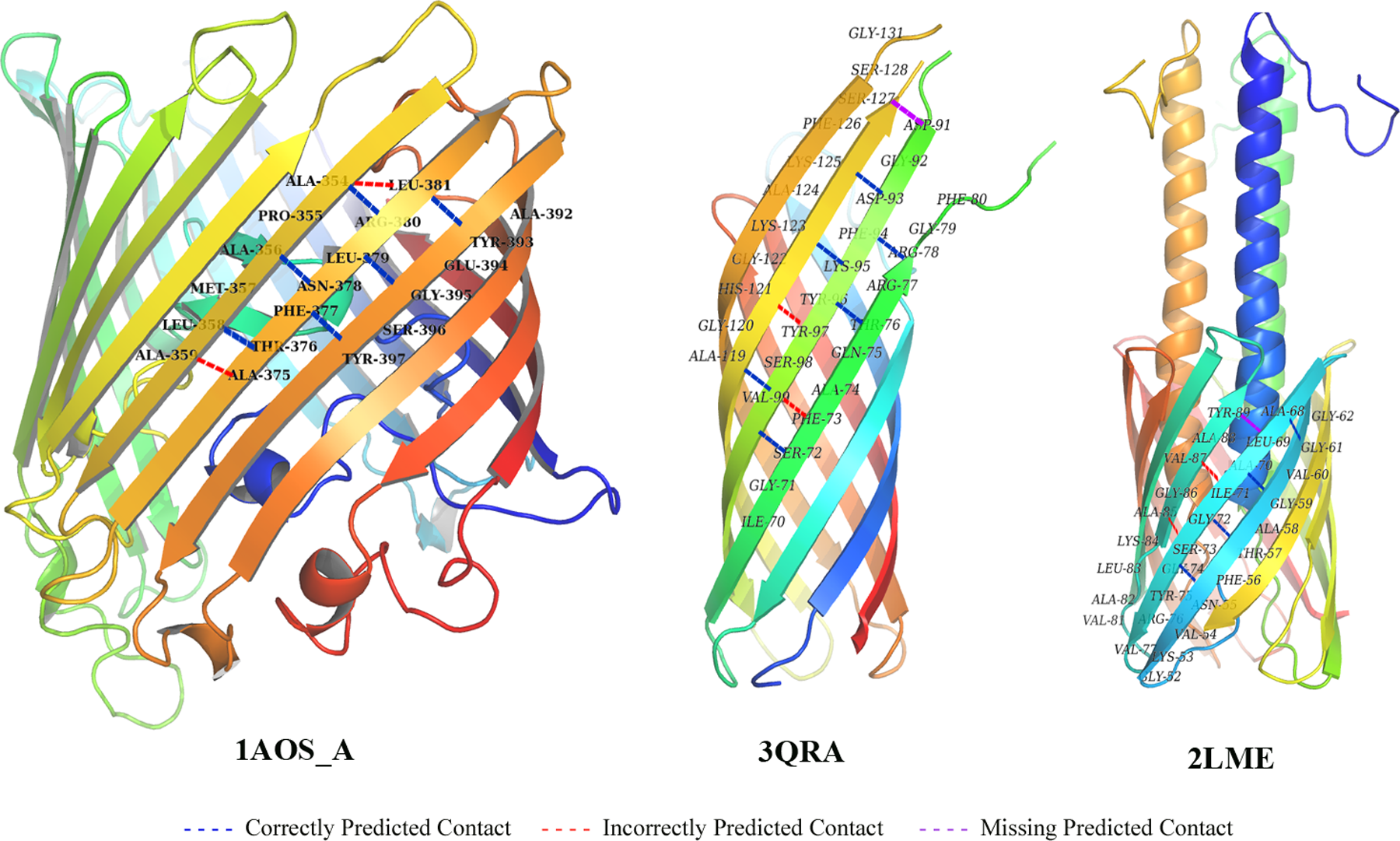
Comparatively, 3QRA is a small OMP with 8 transmembrane segments, and it is less regular in the sequence length of beta sheets and tertiary structure. There were a number of OMPs like 3QRA in the dataset, the corresponding topology predictions were less accurate than those more regular ones, which led to slightly inefficient features used in the contact prediction. It was observed in Figure 4 that the inter-barrel contacts appeared absent sometimes in 3QRA. They should exist according to the same way of 1A0S_A; however, incorrect predictions were made between these residue pairs. At the same time, a contact between sequence position 91 and 127 was missed to prediction, mainly because this contact is close to the end of the two sheets. The topology prediction did not count them in the transmembrane segments, but evolutionary covariation could not determinate whether they have contact by itself. Therefore, the accuracy of contact prediction for 3QRA was a little decreased in the topology feature.
2LME is a special OMP compared to the others, its beta-barrel is not self-circled by one continuous amino acid sequence, but composed of three domains, with both helix and sheet inside the membrane. Meanwhile, 2LME belongs to the GSPII I/J protein-like protein family classified according to CATH. There are a total of six similar structures of 2LME in the family, five of them are not outer membrane proteins, and the other one is not in our non-redundant dataset. Consequently, 2LME is a scarce type of OMP with special inter-barrel residue contacts. From the example, more incorrect and missing contact predictions appeared in the transmembrane segments that have incomplete sheets (e.g., the sheet from sequence position 81 to 89). Another important factor: there exist protein structures inside the beta-barrel of 2LME, which is a vital difference compared to the common OMPs. The residue in these structures will critically change the tertiary structure of beta-barrel. Additionally, the SVM model could not be well trained for such few samples. Consequently, inter-barrel contact prediction accuracy of 2LME is lower than the average level.
4. Conclusions
For the purpose of improving the OMP structure prediction, we implemented an inter-barrel residue contact prediction method called OMPcontact. Observed from the testing results, residue evolutionary covariation can partly represent the residue contact among the residues in transmembrane segments of OMPs, but it also depended on the evolutionary covariation method which had been used. Here, EVFOLD and MI methods were found efficient for OMPs. Topology features were proved impactful, working with evolutionary covariation feature. The topology prediction accuracy will also affect the performance of OMPcontact. Fortunately, most OMPs derived good topology predictions to support the contact prediction.
It is certain that inter-barrel contact would achieve even higher prediction accuracy if topology methods improve their performance in predicting locations of transmembrane segments. At the same time, the OMP lipid layer accessibility, and residue evolution conservation played roles in restricting prediction according to the right way from different perspectives. The test results showed that our method generated the prediction score that can efficiently describe the possibility of corresponding residue pair having contact. Therefore, OMPcontact can be expected to improve the OMP tertiary structure prediction.
Footnotes
Acknowledgments
This work is partially supported by the National Natural Science Foundation of China under Grants No. 61402098 and 61373051, the Jilin Scientific and Technological Development Program of China No. 20140520072JH, 20140204004GX, and 201201048, the Basic Scientific Research from Jilin University (Grant No. 450060481090), and US National Institutes of Health (Grant No. RO1-GM100701).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.