
Abstract
Abstract
Genomics has been used with varying degrees of success in the context of drug discovery and in defining mechanisms of action for diseases like cancer and neurodegenerative and rare diseases in the quest for orphan drugs. To improve its utility, accuracy, and cost-effectiveness optimization of analytical methods, especially those that translate to clinically relevant outcomes, is critical. Here we define a novel tool for genomic analysis termed a biomedical robot in order to improve phenotype prediction, identifying disease pathogenesis and significantly defining therapeutic targets. Biomedical robot analytics differ from historical methods in that they are based on melding feature selection methods and ensemble learning techniques. The biomedical robot mathematically exploits the structure of the uncertainty space of any classification problem conceived as an ill-posed optimization problem. Given a classifier, there exist different equivalent small-scale genetic signatures that provide similar predictive accuracies. We perform the sensitivity analysis to noise of the biomedical robot concept using synthetic microarrays perturbed by different kinds of noises in expression and class assignment. Finally, we show the application of this concept to the analysis of different diseases, inferring the pathways and the correlation networks. The final aim of a biomedical robot is to improve knowledge discovery and provide decision systems to optimize diagnosis, treatment, and prognosis. This analysis shows that the biomedical robots are robust against different kinds of noises and particularly to a wrong class assignment of the samples. Assessing the uncertainty that is inherent to any phenotype prediction problem is the right way to address this kind of problem.
1. Introduction
D
2. The Phenotype Prediction Problem
The primary objective of phenotype discrimination is to define sets of genes/probes that optimally differentiate between populations expressing or not expressing a particular phenotype such as disease risk, drug responsiveness, or medication toxicity. This concept can be useful in identifying biological and molecular pathway differences between normal and cancer cells, or investigating drug mechanisms of action for a certain type of disease.
We built a conceptual model that related different genes/probes to class prediction (phenotype) as a nonlinear classification problem, since the classifier and the genetic features that serve to achieve an optimum prediction of the phenotype are unknown. Therefore, as a first step a given type of classifier (nearest-neighbor, neural networks, support vector machines, etc.) should be built ad-hoc to relate the genetic features to the observed phenotype classes. The classification problem of phenotype discrimination does not need necessarily to be binary, it could be multiclass. This can be considered as the first source of uncertainty in the phenotype prediction problem, since the perfect classifier is a priori unknown.
First we start with a set of expressions of n genes/probes for a set of m samples whose phenotype classes are defined, usually by medical expert annotation. This information is typically organized in the expression matrix
Importantly, not all the genes/probes provide useful information to the phenotype prediction inverse problem. These extraneous genes are noisy and can be analytically disruptive. Fortunately, it is possible to discard irrelevant features, that is, those genes that do not provide any useful information for the phenotype discrimination, since these features introduce ambiguity in the classification. The relevant genes would be defined as the ones that minimize a given target function
where • First, several equivalent genetic signatures exist that explain the phenotype class equally well or have a similar predictive accuracy. This is known as the ill-posed character of the phenotype classification problem. Thus, we can apply the parsimony principle to identify small-scale signatures by introducing the concept of redundancy. Given a genetic signature • Second, the ill-posed character of the classification is due to the high underdetermined character of the inverse problem involved, since the number of samples • Third, genomic data is notorious for containing noise that has historically contributed to issues around reproducibility, especially as related to gene/clinical phenotype relationships. Similarly, genomic noise also impedes accurate mechanistic conclusions by partially falsifying biological pathways. There are two main sources of noise: • First, noise in the genes’ expressions that is introduced by the process of data filtering and measurement. The observed genetic expression of a sample, • Second, noise in the class assignment
It is straightforward to show that both kinds of noise induce a modeling error in the classifier. Therefore, in the presence of these types of noise (
3. Biomedical Robots
A biomedical robot is a set of algorithms derived from applied mathematics, statistics, and computer science that are capable of dynamically analyzing high-dimensional data, discovering knowledge, generating new biomedical working hypothesis, and supporting medical decision making with its corresponding uncertainty assessment. In this definition the data does not need necessarily to be of the same type; that is, several types of data could be used for decision-making purposes. In the present case the data come from microarray differential expression analysis, between individuals that develop the illness and others that do not.
Generating a new working hypothesis in the present case includes the analysis of biomarkers and mechanisms of action involved in the illness development, and finding existing drugs that could target the main actionable genes. Also, a benefit of this approach could be the design of intelligent systems to support medical doctors/researchers in the decision-making process of new incoming uncatalogued samples to decide questions relative to their diagnosis, treatment, and prognosis before any decision is taken. These techniques can help (de Andrés-Galiana et al., 2015, 2016) for instance in segmenting patients with respect to response to treatment (deAndrés-Galiana et al., 2015) and also to drug response, to predict the development of induced toxicities (Saligan et al., 2014), to infer the possible surgical risk, etc., among many different applications that we can imagine.
Figure 1 shows a conceptual scheme of the biomedical robot concept. From a training data set we built
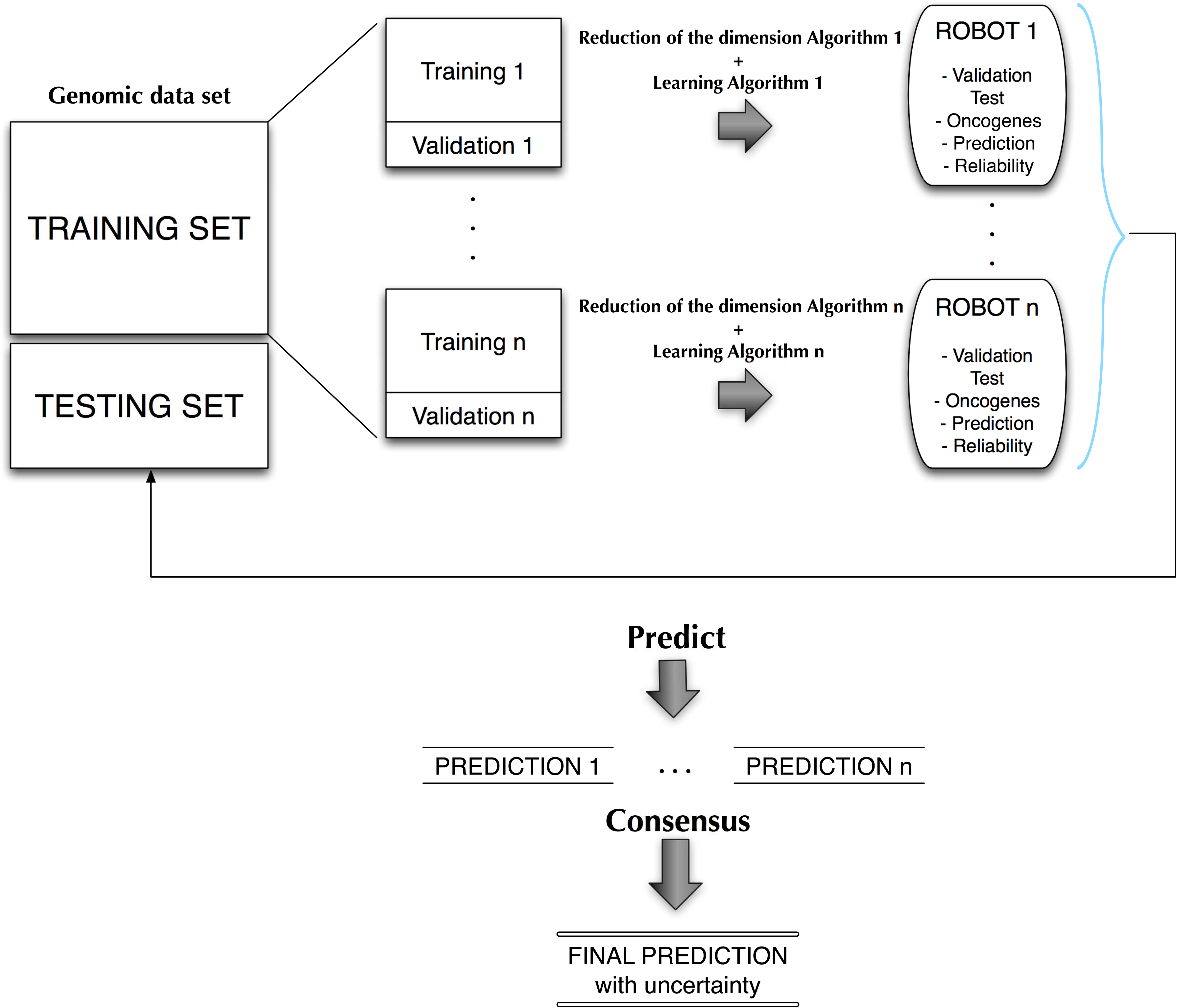
Conceptual scheme for the design of biomedical robots.
It is important to remark that we are not interested in building a black-box methodology, but also being able of inferring the mechanisms of action and the genetic and biological pathways that are involved. The final decision approach is as follows: Given a new incoming sample, each of the equivalent robots will perform a prediction. A final prediction with its uncertainty assessment will be given using all these predictions via a consensus strategy such as majority voting. This approach has been used by Fernández-Martínez and Cernea, (2014) in a face recognition problem obtaining very high stable accuracies. Ensemble classification and majority vote decisions are based on Condorcet's jury theorem, which is a political science theorem about the probability of a given group of individuals arriving at a correct decision. In the context of biomedical robots and ensemble learning, it implies that the probability of being correct for a majority of independent voters is higher than the probability of any of the individual voters, and tends to one when the number of voters (or weak classifiers) tends to infinite. In this case the weak classifiers are any of the biomedical robots of the ensemble that have a high predictive accuracy. These classifiers are guaranteed to be independent since they use different high discriminatory genetic signatures, measured by their corresponding discriminatory power.
Several methods exist to assign the discriminatory power of the genes: fold-change (Schena et al., 1996), Fisher's ratio (Fisher, 1936), entropy (Shannon, 1948), mutual information (Quinlan, 1993), significance analysis of microarrays (SAM) (Tusher et al., 2001), percentile distance between statistical distributions (deAndrés-Galiana et al., 2015), etc. Generally speaking, high-discriminatory genes are those that have very different distributions in both classes (in a binary problem) and whose expression remains quite stable or homogeneous within each class.
The algorithm used in this article is similar to the one that was introduced in Saligan et al. (2014) and (deAndrés-Galiana et al., 2015) and consists of several steps (see Fig. 1):
1. Applying several filter feature selection methods to find different lists of high discriminatory genes. 2. Establishing the predictive accuracy of these lists of genes using a leave-one-out-cross-validation (LOOCV) cost function via a k-nearest-neighbor (k-NN) classifier. Others classifiers could also be used. This sampling procedure of the phenotype prediction uncertainty space aims at obtaining from these lists different biomedical robots with their corresponding predictive accuracy. For that purpose we can use backward feature elimination and/or random sampling methodologies. 3. Selecting robots above a certain predictive accuracy (or below a given error tolerance) and performing the consensus prediction through majority voting.
According to the definitions stated in (1), (2), (3), and (4) we can formally define a biomedical robot as the set of classifiers:
whose predictive error (the number of misclassified samples) is lower than a given bound
In this article we apply this concept to the analysis of three kinds of diseases: cancer (CLL), neurodegenerative (ALS), and rare diseases (IBM-PM). Although the concept is theoretically correct before applying it to these datasets, we have analyzed its robustness against different types of noises using synthetic microarrays. This analysis helped us to extract interesting conclusions regarding the interpretation of the results obtained for real datasets.
3.1. Noise sensitivity analysis
We have generated different synthetic data sets using three types of noise: additive Gaussian noise, lognormal noise, and noise in class assignment. These last two types belong to the category of non-Gaussian noise, since they are multiplicative and systematic random noises. The method consists in building a synthetic dataset with a predefined number of differentially expressed discriminatory genes, and subsequently introducing different types of noise, and determining the predictive accuracy (Acc) as a function of the number of applied robots (#R). The synthetic dataset was built using the OCplus package available for The Comprehensive R Archive Network (Pawitan and Ploner, 2015).
Table 1 shows the results obtained for the sensitivity analysis.
The following information is given: δ, the percentage of noise introduced; Acc, the mean LOOCV predictive accuracy; P, the precision of the selection using the union of all the genes found by the robots; and #R, the number of robots applied in the consensus strategy.
where • The precision P keeps quite stable when noise in class assignment is increased. This result is very interesting since the biomedical robots are able to find the differentially expressed genes when the noise in class assignment is introduced. In the case of Gaussian noise the precision is very high for noise levels less than 5%. The worst result was obtained when multiplicative noise is added to the expressions. The fact that the precision gradually decreases when noise in the expression increases implies that some of the biological pathways that are inferred might be partially falsified. Therefore, any filtering step that is usually performed in the microarray data will have important consequences with respect to the pathway analysis. Future research will be devoted to this important subject. • The mean predictive accuracy (Acc) systematically decreases when a higher level of noise is added to the class assignment vector, and is very stable when Gaussian and non-Gaussian noises are added to the expression data, meaning that the biomedical robots are robust in terms of accuracy with respect to the presence of noise in the expressions. This result also suggests that noise acts as regularization with respect to the accuracy in the prediction as it has been theoretically proven by Fernández-Martínez et al. (2014a, b) in inverse problems. It can be also concluded that if the biomedical robots are unable to improve the accuracy of the best prediction, the dataset must have some wrong class assignment that prevents achieving a perfect classification. Another possibility is that parameterization of the samples is incorrect; in the present case that would mean that none of the genes that have been measured bring enough information to achieve a good phenotype discrimination.
3.2. Chronic lymphocytic leukemia
B-cell chronic lymphocytic leukemia (CLL) is a complex and molecular heterogeneous disease, being the most common adult leukemia in western countries. In our cohort, DNA analyses served to distinguish two major types of CLL with different survival times based on the maturity of the lymphocytes, as discerned by the immunoglobulin hyeavy chain variable-region (IgVH) gene mutation status (Ferreira et al., 2014). In this first example we had at our disposal a microarray dataset consisting of 163 samples and 48807 probes.
The best robot predicted the IgVH mutational status with 93.25% accuracy using small-scale signature composed by 13 genes: LPL (2 probes), CRY1, LOC100128252 (2 probes), SPG20 (2 probes), ZBTB20, NRIP1, ZAP-70, LDOC1, COBLL1, and NRIP1.
Table 2 shows the results of applying the methodology of biomedical robots to this problem. In this case the highest prediction accuracy obtained by the set of biomedical robots equal the accuracy provided by the best robot (93.25%). This result implies that some samples are behavioral outliers or might be misclassified. This happened with 11 samples that are identified in the PCA graphic in two dimensions (Fig. 2) using the genetic signature composed of these 13 genes. It can be observed how the classification in this reduced set of genes becomes almost linearly separable; while using all the genetic information that we have at our disposal the classification is nonlinear. Therefore, as an important conclusion we can affirm that reducing the dimension to the set of discriminatory genes helps to linearize the phenotype classification problem.
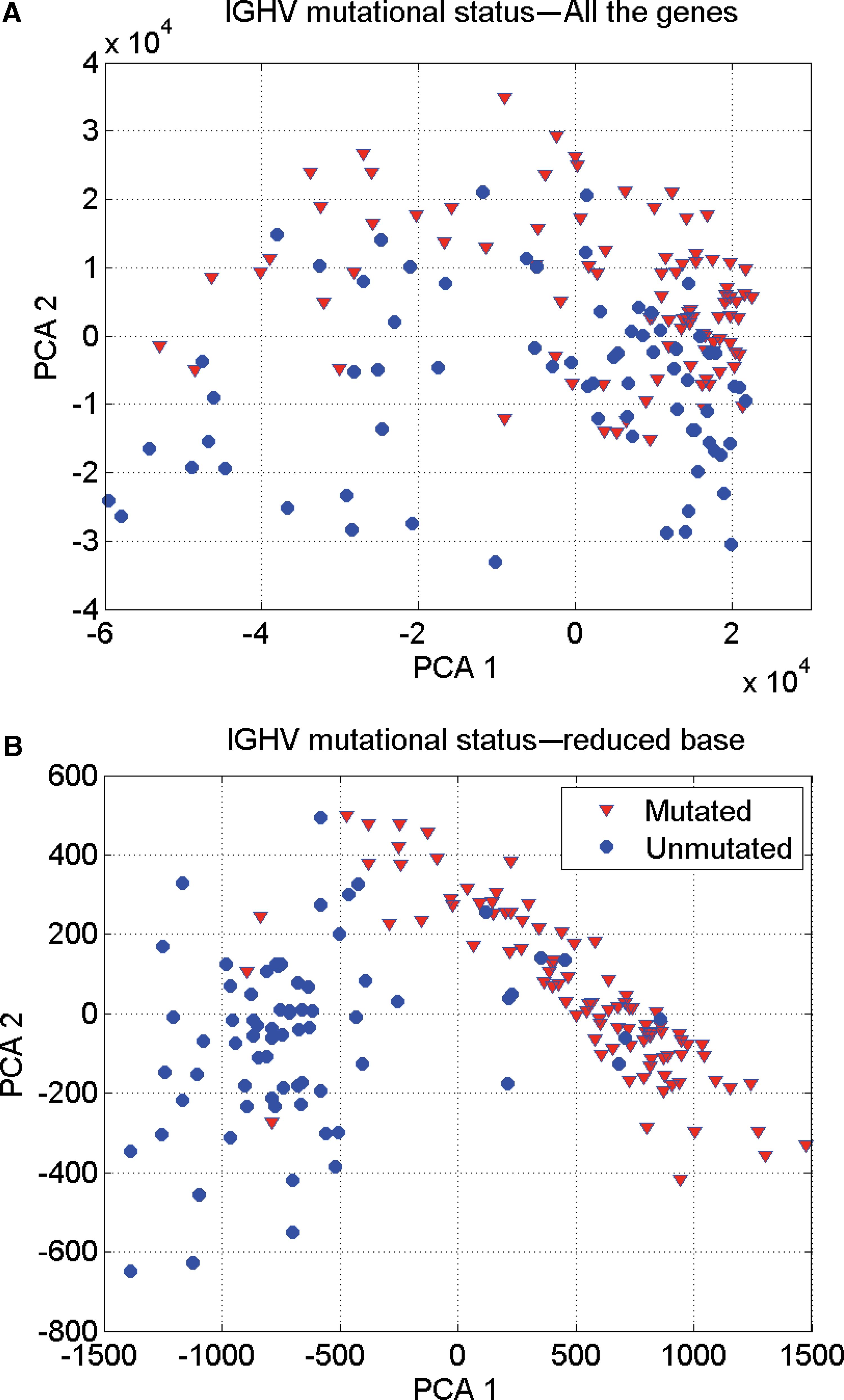
IgVH classification in CLL:
Figure 3 also shows the correlation network of the most discriminatory genes of the CLL-IgVH mutational status found in this analysis. This is an interesting tool to understand how the most discriminatory genes regulate the expression of other genes involved in different biological pathways. The head of the graph is the gene that has the highest discriminatory power LPL. It can be observed as one main network associated with ZBTB20.
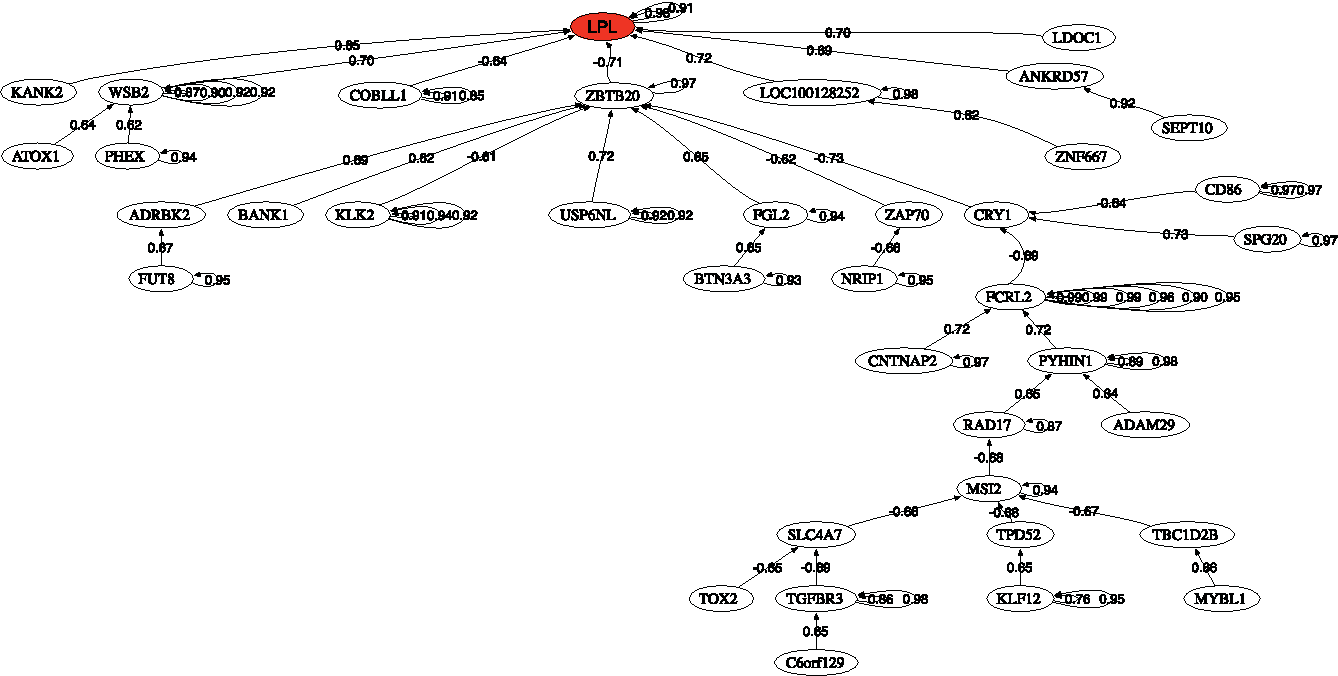
Correlation network for IgVH mutational status in chronic lymphocytic leukemia.
Finally the pathway analysis deduced from the biomedical robots has revealed the importance of the ERK signaling super pathway that includes ERK signaling, ILK signaling, MAPK signaling, molecular mechanisms of cancer, and rho family GTPases pathway. These pathways control proliferation, differentiation, survival, and apoptosis. Also, other important pathways found were allograft rejection, the inflammatory response pathway, CD28 costimulation, TNF-alpha/NF-kB signaling pathway, akt signaling, PAK pathway, and TNF signaling. The presence of some of these pathways suggests viral infection as a possible cause for CLL.
3.3. Inclusion body myositis and polymyositis
Myositis means muscle inflammation and can be caused by infection, injury, certain medicines, exercise, and chronic disease. Some of the chronic, or persistent, forms are idiopathic inflammatory myopathies, whose cause is unknown. We have modeled the inclusion body myositis /polymyositis (IBM/PM) dataset published by Greenberg et al. (2005). The data consisted of the microarray analysis of 23 patients with IBM, 6 with PM, and 11 samples corresponding to healthy controls. The best robot perfomed the classification of the IBM+PM vs. control, obtaining a predictive accuracy of 97.5% using a reduced base with only 17 probes. The genes belonging to the highest predictive small-scale genetic signature are HLA-C (three probes), HLA-B (four probes), TMSB10, S100A6, HLA-G, STAT1, TIMP1, HLA-F, IRF9, BID, MLLT11, and PSME2. It can be observed in the presence of different HLA-x genes of the major histocompatibility. Particularly the function of the gene HLA-B would explain alone the genesis of IBM: “HLA-B (major histocompatibility complex, class I, B) is a human gene that provides instructions for making a protein that plays a critical role in the immune system. HLA-B is part of a family of genes called the human leukocyte antigen (HLA) complex. The HLA complex helps the immune system distinguish the body's own proteins from proteins made by foreign invaders such as viruses and bacteria.”
Table 2 shows the results using the biomedical robots methodology. In this case we are able to hit 100% of the samples with two robots, improving the results of the best robot. The analysis of biological pathways has revealed the importance of viral infections, mainly in IBM patients: allograft rejection, influenza A, class I MHC mediated antigen processing and presentation, Staphylococcus aureus infection, interferon signaling, immune response IFN alpha/beta signaling pathway, phagosome, tuberculosis, cell adhesion molecules (CAMs), Epstein-Barr virus infection, and TNF signaling. Several viral infections appeared in this list. Interestingly, it has been found that 75% of the cases of viral myosytis are due to Staphylococcus aureus infection (Fayad et al., 2007).
Figure 4 shows the correlation network of the most discriminatory genes found in this analysis. It can be observed as the presence of one main dense network involving different HLA-X genes. Among its related pathways are ERK signaling and apoptosis pathway. GO annotations related to this gene include calcium ion binding and cysteine-type peptidase activity.
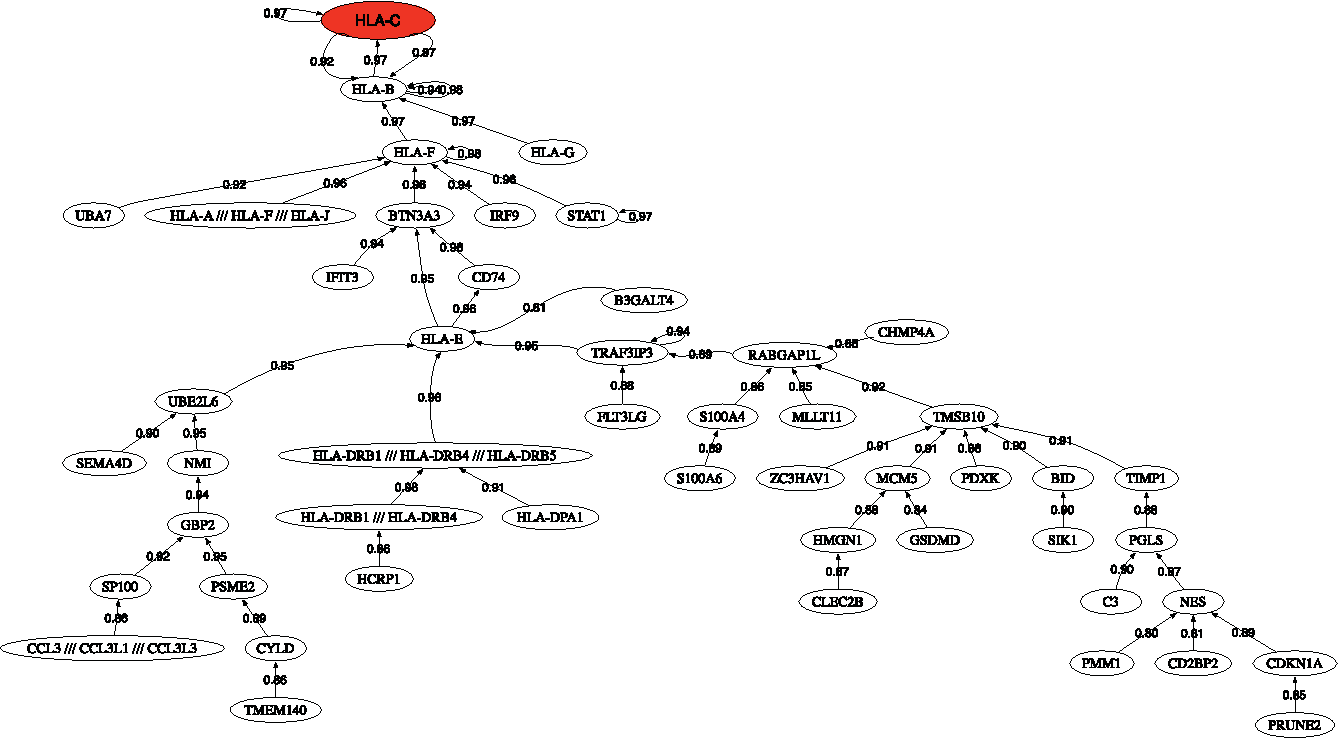
Correlation network for inclusion body myositis/polymyositis.
Figure 5A shows the PCA projection for the IBM+PM versus control samples using the optimum reduced base. It can be observed that the separability is almost perfect and only one PM sample that is close to the control samples might be misclassified. This graphic also explains that this basis set is not optimum to perform the classification of IBM vs. PM. This separability can be achieved with 100% accuracy using a reduced base composed by the following genes: RHOBTB2, MT1P2, FBXL8, HIF3A, C17orf101, RPL12, RBM19, MT1G, WT1-AS, HEXIM1, NQO2, ENOSF1, ADRM1, EIF5A, CSF2RA, CPLX3 /// LMAN1L, C10orf95, NFIC, and POLR2J2. The main pathways involved in the IBM vs. PM phenotype differentiation is: FOXA1 transcription factor network, O2/CO2 exchange in erythrocytes, methotrexate pathway, drug induction of bile acid pathway, bile secretion, and statin pathway. Figure 5B shows the PCA graphic of the IBM vs. PM classification, and how this separability can be achieved.
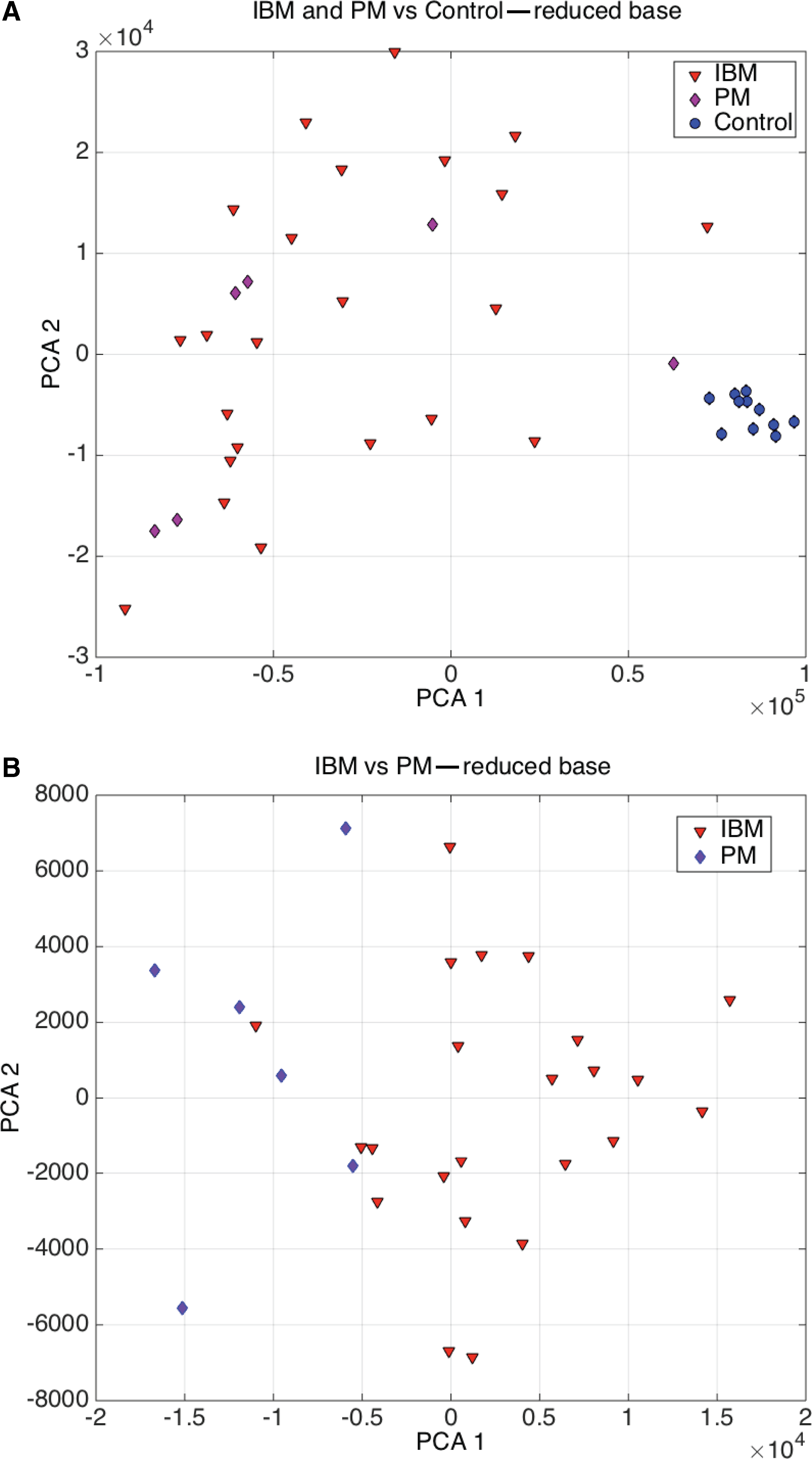
Classification of IBM, PM, and Control:
3.4. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease that's characterized by stiff muscles, muscle twitching, and gradually worsening weakness. Between 5 and 10% of the cases are inherited from a relative, and for the rest of the cases, the cause is still unknown (NINDS, 2013). It is a progressive disease in which the average survival from onset to death is 3 to 4 years, and most of the deaths are caused by respiratory failure. There is no cure yet.
We reinterpreted the dataset published by Lincecum et al. (2010) consisting of 57 ALS cases and 28 healthy controls. The best result yields an accuracy of 96.5% with small-scale signature involving the following genes: CASP1, ZNF787, and SETD7. Table 2 shows the results of applying this methodology to this problem. The biomedical robots in this case did not improve this prediction. The pathway analysis has revealed the importance of the GPCR pathway, RhoA signaling pathway, EPHB forward signaling, ephrinA-EphR signaling, EBV LMP1 signaling, and regulation of microtubule cytoskeleton. These pathways have different important signaling roles and suggest a possible link to the Epstein-Barr virus (EBV).
Figure 6 shows the correlation network of the most discriminatory genes found in this analysis. The head of the network is the CASP1 that is connected to MAP2K5 through ZNF3 and LUC7. MAP2K5 acts as a scaffold for the formation of a pathway that seems to play a critical role in protecting cells from stress-induced apoptosis, neuronal survival, cardiac development, and angiogenesis. Also DCAF8 has been associated with neuropathies.
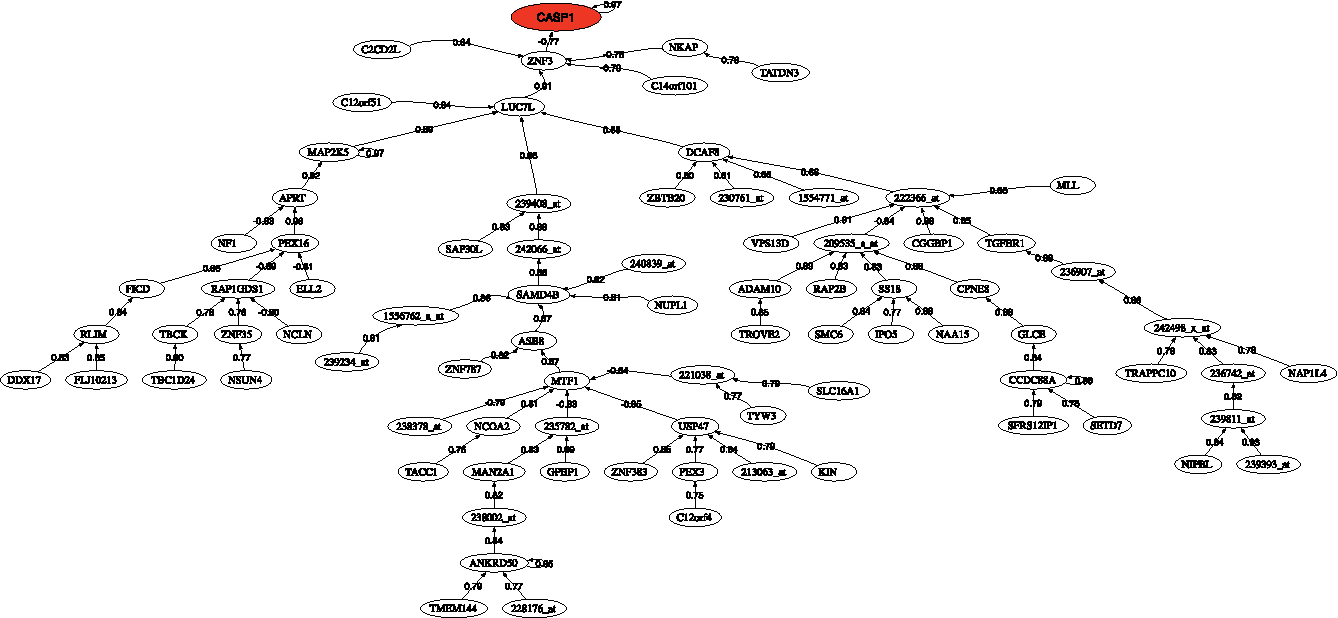
Correlation network for amyotrophic lateral sclerosis. Probe names are used when gene names are unknown.
Figure 7 shows the PCA graphic for the ALS vs. control samples. It can be observed that the accuracy of the classification could be easily improved by discarding 5–6 control samples that lie very close to the border defined by the ALS samples. Also it can be observed that one ALS sample is clearly a behavioral outlier.

PCA graphic for ALS versus control samples.
4. Conclusion
In this article we have introduced the concept of biomedical robots, performing its sensitivity analysis to different kinds of noises, and showing its application to the analysis of cancer, as well as rare and neurodegenerative diseases. The concept of the biomedical robot is based on exploring the uncertainty space of the phenotype classification problem involved and using the structure of the uncertainty space to adopt decisions and inferring knowledge. The synthetic dataset modeling has shown the robustness and stability of this methodology, particularly to class assignment noise. The presence of high noise levels in expressions might falsify the biological pathways that are inferred. Nevertheless, the predictive accuracy remains very high. Finally, we have shown the application of this novel concept to three different illnesses: CLL, IBM-PM, and ALS, proving that it is possible to infer at the same time both high discriminatory small-scale signatures and the description of the biological pathways involved. We have shown that referring to the set of most discriminatory genes these classification problems become linearly separable. Generally speaking in the three cases no high class assignment errors have been detected, being CLL the case where more samples (11) have been found to be behavioral outliers. The pathway analyses revealed in the three cases a possible link to viral infections and served to identify actionable genes and drug targets. The methodology shown in this article is not computationally very expensive, since all the simulations shown in this article were done with a personal computer in real time (several minutes).
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist. Enrique J. de Andrés was supported by the Spanish Ministerio de Economía y Competitividad (grant TIN2011-23558).