
Abstract
Abstract
This article presents an efficient heuristic for protein folding. The protein folding problem is to predict the compact three-dimensional structure of a protein based on its amino acid sequence. The focus is on an original integer programming model derived from a platform used for Contact Map Overlap problem.
1. Introduction
A
In this study, we consider the hydrophobic–hydrophilic (HP) model for a two-dimensional (2D) square lattice. The first HP model was introduced by Dill (Dill,1985; Dill et al., 1995; Yoon, 2006). The 20 types of amino acids ingredients of a protein are classified as hydrophobic (H) or polar (P) by degree of hydrophobicity of amino acids. Then the HP model simplifies the protein-folding problem by considering only two types of amino acids: H and P. Lattice protein models locate each amino acid on a point of a 3-D cubic lattice, and in order to maintain the connectivity of the protein, amino acids that are adjacent in the protein's sequence must also occupy adjacent lattice points.
Proteins fold in three dimensions. However, scientists often use a 2-D model instead of the 3-D model to test their algorithms (i.e., they use the square lattice instead of the cubic lattice), and attempt to extend the 2-D model into 3-D. In this report, we will focus on one specific type of protein-folding model that can be described in the following manner:
Maximize the number of H–H contacts subject to:
1. Assignment: Each amino acid must occupy one lattice point, 2. Non-overlapping: No two amino acids may share the same lattice point, 3. Connectivity: Every two amino acids that are consecutive in the protein's sequence must also occupy adjacent lattice points.
The objective function of this model is to maximize the number of H–H contacts, which is the number of adjacent (in the lattice) hydrophobic amino acids (Chandru et al., 2004).
This problem (even for a square lattice) is proven NP-complete (Alberts et al., 1998; Jiang and Zhu, 2005; Ahn and Park, 2010). Therefore, finding a good heuristic is a challenge partially overcome by the algorithm described below (Duan and Kollman, 2001; Michalewicz and Fogel, 2004).
2. Mathematical Model
The mathematical model is based on an upright square lattice with a fixed size (Carr et al., 2003; Istrail et al., 2000). Such a lattice is conveniently presented as a m × m checkerboard with the neighborhood of each white (black) square—the four black (white) surrounding squares. Let now Gc = (Vc, Ec) be a graph with Vc = {1,2,.…,m2}, where node i corresponds to the i-th square (under an arbitrary numeration of the squares) and the edge (i, j) ∈ Ec if i and j are neighbors. Conveniently, the four edges incident with a given vertex i will be labeled with: u,d,l,r for the up, down, left, and right surrounding squares. (The border squares, resp. nodes, are of smaller than 4 degrees.) The simple paths (each node is visited at most once) in Gc are called a
Let S be a sequence of n letters on {0, 1} alphabet (0-for P and 1-for H). Let GS = (VS, ES) be a graph associated with S with a node set VS = {1, 2, …, n} and (i, j) ∈ ES if and only if |i – j| ≥ 2 and S[i] = S[j] = 1. Let G = GS ∪ Gc be a complete bipartite graph with node set VS ∪ Vc. The matching (in this case, one-to-one mapping of VS to Vc) M = {e1, e2, …, en} with |M| = n is
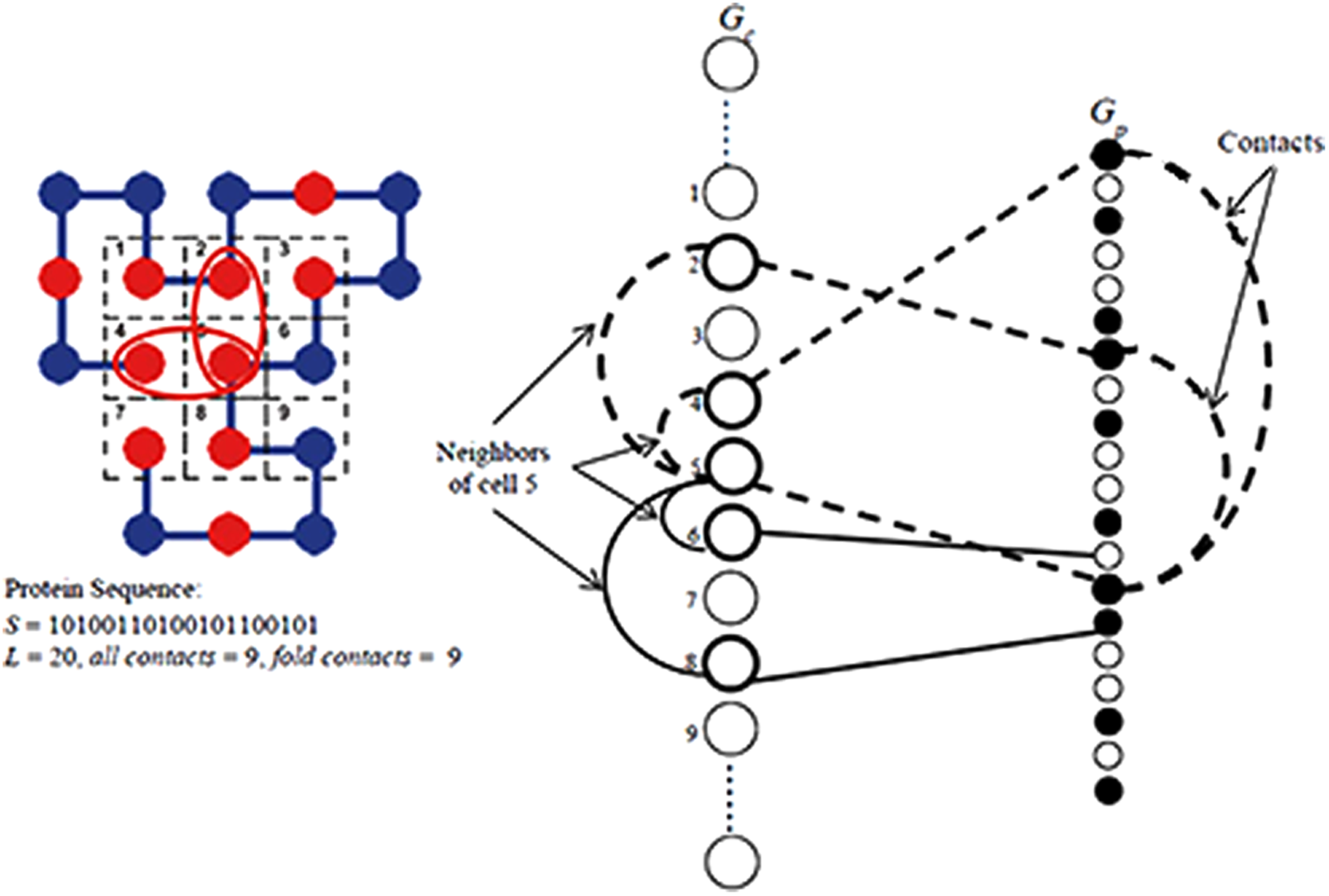
One-to-one mapping of VS to Vc.
Then the problem of finding the optimal folding over up right square lattice is:
Square folding problem (SFP). For
Converting the HP problem as an optimization problem on graphs allows for building various integer programming models. Most of them involve introducing binary variables, say xik for modeling the feasible matchings from above as 0-1 solutions to the simple linear constraints:
(assignment)
(non-overlapping)
The objective function could be expressed by: linearization of zijkl = xikxjl and/or by partitioning the sum of z in sub-sums in different ways. We will not present here any possible integer programming models, since our goals do not involve solving such models, but for sake of completeness, we add the following constraints to finish modeling the self-avoiding paths:
where n(i) ≔ {i − 1, i + 1, i − m, i + m} is the set of neighbors of the i-th square under the row-wise numbering of the checkerboard squares.
An easily derivable bound to v could be obtained by the following observation: Let ODD be the set of odd i, s.t. S[i] = 1 and EVEN be the set of even i, s.t. S[i] = 1. Since w.l.o.g. even elements of S are assigned to black squares of the checkerboard, and odd elements to the white ones, then zikjl could be equal to 1 only for even-odd couples i, j. Then C2D(S) = 2*min{|ODD|, |EVEN|} is obviously an sharp upper bound to v. (If S starts or ends with 1 this bound should be increased by 1 or 2.)
Getting back to the HP folding problem and its conversion to a problem of finding matching that maximizes the number of overlapping edges, one could find a lot of similarity with another problem known as Contact Map Overlap (CMO). The problems in this class were well solved by applying lagrangean relaxation techniques to the corresponding integer programming models (Malod-Dognin et al., 2008; Yanev et al., 2008). Unfortunately, applying such technique to the SFP is prevented by the bad cutting properties of the bounds obtained by the LP relaxation as well and Lagrangean dual bound which coincide with trivially computable C2D(S). These bounds (bound) are non-improvable deep in the branch and bound tree and thus the direct application of integer programming solvers is still questionable. The heuristic, given below, is a reasonable candidate to overcome these difficulties and due to the new insight on HP problems as a kind of contact map overlap, it could be applied to a broader class of optimization problems.
3. Heuristic Algorithm
Let i = {2, 3, …, n } = ∪
k
Si Si∩Si+1 = ∅ and
3.1. Algorithm GREEDYLIKE
4. Computational Experiments
The computational runs illustrated below are for Python realization of the algorithm on an Intel Core i5 430M (2.26 GHz, 3 MB L3 cache), 4 GB RAM laptop. As a first demonstration of its capabilities, we borrow some figures from Istrail and Lam (2009) for a small (36 amino acids) human protein: PPPHHPPHHPPPPPHHHHHHHPPHHPPPPHHPPHPP. Figure 2 shows the fold obtained by our algorithm. This fold is close to the optimal one given on Figure 3c and much better than the folds obtained by approximate algorithms, shown on Figure 3a and b.

Protein folding with length 36 amino acids. The number of obtained contacts is 13.

More extensive results for proteins of different lengths are listed in Table 1. The comparative results for the first eight benchmarks are shown in Table 2 (Toma and Toma, 1996; Chen and Huang, 2005; Ahn and Park, 2010). The column segment size in Table 1 gives the value of the only parameter of the algorithm (see len(Si) in the previous section). Since the function PATHFINDER is a kind of total enumeration, increasing its value above 12 could consume a prohibitively large time.
As we can see from Table 2, the GREEDYLIKE algorithm finds solutions that are very close to optimal. Even for some sequences, we find the optimal solution. The output for much longer proteins is not given here, since there is nothing with which to compare.
The resulting folds for sequences with length 24, 36, and 60 amino acids are provided on Figure 4.

Folding of proteins with lengths:
5. Conclusion
Computational experiments show that the idea of decomposing the problem into tractable subproblems works well for arbitrary long protein sequences. What could be added to the idea is to extend the size of the subproblems by replacing the PATHFINDER function with stronger solvers. Ahead of us stands the challenge to implement the algorithm on a larger segment size by changing PATHFINDER with an exact algorithm based on an integer programming approach. In fact, by using advanced integer programming models, we were able to solve to optimality all instances from Table 1, but there are still problems with skipping the length size 100 barrier. Also, we can improve the quality of folds obtained from the proposed method by adapting to other lattice models (including 3D lattice) or insertion of other techniques for analysis of protein structure.
We note that coding of this algorithm is not complicated, and thus can be easily applied in practice.
Footnotes
Acknowledgments
This work is partially supported by the project of the Bulgarian National Science Fund, entitled: “Bioinformatics research: Protein folding, docking and prediction of biological activity,” code NSF I02/16, 12.12.14.
Author Disclosure Statement
The authors declare that no competing financial interests exist.