
Abstract
Abstract
High-throughput experimental techniques have been producing more and more protein–protein interaction (PPI) data. The PPI network alignment greatly benefits the understanding of evolutionary relationship among species, helps identify conserved subnetworks, and provides extra information for functional annotations. Although a few methods have been developed for multiple PPI network alignment, the alignment quality is still far from perfect, and thus, new network alignment methods are needed. In this article, we present a novel method, denoted as ConvexAlign, for joint alignment of multiple PPI networks by convex optimization of a scoring function composed of sequence similarity, topological score, and interaction conservation score. In contrast to existing methods that generate multiple alignments in a greedy or progressive manner, our convex method optimizes alignments globally and enforces consistency among all pairwise alignments, resulting in much better alignment quality. Tested on both synthetic and real data, our experimental results show that ConvexAlign outperforms several popular methods in producing functionally coherent alignments. ConvexAlign even has a larger advantage over the others in aligning real PPI networks. ConvexAlign also finds a few conserved complexes, which cannot be detected by the other methods.
1. Introduction
P
PPI networks can be aligned either locally or globally. Local network alignment methods aim to find conserved subnetworks among a set of input networks (Koyutürk et al., 2006; Ciriello et al., 2012), while global network alignment (GNA) methods maximize the overall match between input networks (Singh et al., 2007, 2008; Kuchaiev and Pržulj, 2011; Patro and Kingsford, 2012; Neyshabur et al., 2013; Todor et al., 2013; Hashemifar and Xu, 2014; Saraph and Milenković, 2014; Vijayan et al., 2015). More methods are developed for pairwise network alignment. With the availability of more PPI networks, it becomes inevitable to align multiple networks. Existing GNA methods such as NetworkBlast-M (Sharan et al., 2005; Kalaev et al., 2008) and GraemLin2.0 (Flannick et al., 2008) are designed for local alignment of multiple networks, whereas others such as IsoRankN (Liao et al., 2009), SMETANA (Sahraeian and Yoon, 2013), NetCoffee (Hu et al., 2014), BEAMS (Alkan and Erten, 2014), and FUSE (Gligorijević et al., 2015) are for GNA. Most methods make use of both sequence similarity and network topology, and are designed for many-to-many alignments.
NetworkBlast-M starts with a set of highly conserved regions and then extends them greedily. GraemLin2.0 integrates phylogenetic information and network topology and then uses a hill-climbing algorithm to generate the alignment. IsoRankN applies IsoRank to compute pairwise alignment scores first and then uses a PageRank-Nibble algorithm to cluster the proteins. SMETANA uses a semi-Markov random walk model to measure similarity between proteins. BEAMS constructs a weighted k-partite graph in which edge weights are derived from protein sequence similarity. NetCoffee applies a triplet approach to compute the edge weights of the k-partite graph. Both BEAMS and NetCoffee apply a heuristic on the k-partite graph to build an alignment. BEAMS fulfills this by greedily merging a set of disjoint cliques whereas NetCoffee does this by applying a simulated annealing method. FUSE applies a non-negative matrix tri-factorization method to compute edge weights of the k-partite graph.
Although a few GNA methods have been developed, GNA is still far from mature, especially for multiple network alignment (MNA). First, many existing methods do not optimize alignment of all proteins simultaneously. They either use a seed and extension or progressive strategy to build an MNA, so they cannot easily fix errors introduced at an early stage. Second, many existing methods do not fare well in detecting proteins that are functionally conserved across multiple (≥4) PPI networks, partially due to their greedy strategy of building an MNA.
This article presents a novel one-to-one algorithm, ConvexAlign, to align multiple PPI networks using a new scoring scheme that integrates network topology, sequence similarity, and interaction conservation score. We formulate GNA problem as an integer program and relax it to a convex optimization problem, which enables us to simultaneously align all the PPI networks, without resorting to the widely used seed and extension or progressive alignment methods. We use an alternating direction method of multipliers (ADMM) method to solve the relaxed convex optimization problem and optimize all the protein mappings together. Experimental results indicate that ConvexAlign outperforms state-of-the-art methods in terms of biological alignment quality and finding conserved complexes.
2. Methods
2.1. Develop a much more sensitive scoring function
Our goal is to find an alignment that maximizes the number of preserved edges and the number of matched orthologous proteins. Our scoring function consists of two items; one for protein similarity and the other for interaction conservation, that is, we score an alignment
where
where
where
2.2. Integer and convex programming formulation
Let M be the number of proteins in all the input PPI networks, that is,
where each block Xij = YiYjT is a binary matrix encoding the mapping between Vi and Vj. That is, Xi,j(vi,vj) = 1, if and only if, vi and vj are aligned (i.e., in the same alignment cluster).
where
where
The nonlinear constraint (5) can be replaced by the following linear inequalities [c.f. Kumar et al., (2008) and Huang et al., (2011)]:
It is easy to prove that (5) implies (7). On the other direction, considering that the coefficient of
where
Finally, by integrating (4), (6), and (8) and Proposition 1 (Supplementary Material), we have the following integer program:
subject to
The key constraint is
2.3. Optimization via convex relaxation
It is NP-hard to directly optimizing (9) since the variables are binary. We first relax
Afterward, we use ADMM to solve the convex relaxationship (10). The basic idea is to augment its Lagrangian dual and iteratively optimize a subset of variables while keeping the others fixed (details in Supplementary Material Section H). Finally, we propose a greedy rounding strategy to convert fractional solution to integral. We collect all the protein pairs with an indicator value
3. Results
We compare ConvexAlign with state-of-the-art methods IsoRankN (Liao et al., 2009), SMETANA (Sahraeian and Yoon, 2013), NetCoffee (Hu et al., 2014), BEAMS (Alkan and Erten, 2014), and FUSE (Gligorijević et al., 2015). We ran SMETANA, NetCofee, and FUSE with their default parameters. For BEAMS and IsoRankN, we set three values for their parameter α = {0.3, 0.5, 0.7}.
Test data: We use the PPI networks of Homo sapiens (human), Saccharomyces cerevisiae (yeast), Drosophila melanogaster (fly), Caenorhabditis elegans (worm), and Mus musculus (mouse) taken from IntAct (Kerrien et al., 2011). The network properties are shown in Table 1.
We also use NAPAbench (Sahraeian and Yoon, 2012), an extensive alignment benchmark that contains synthetic PPI network families generated by three different network models: crystal growth (Kim and Marcotte, 2008), duplication-mutation-complementation (Vázquez et al., 2003), and duplication with random mutation (Solé et al., 2002). We use the eight-way alignment data set, which contains 3 network families, each with 8 networks of 1000 nodes. The eight-way alignment data set simulates a network family of closely related species, so this benchmark has very different properties as the aforesaid five real PPI networks.
Alignment quality measures: We evaluate MNA quality using several topological and functional consistency metrics proposed in different studies. These metrics are defined in Supplementary Material Section F. Functional consistency measures, however, are more important than topological measures since one of the important applications of network alignment is to functional annotation transfer. For topological analysis of the output clusters, we use c-coverage, ratio of conserved interaction (RCI), and conserved interaction quality (CIQ). A multiple alignment with a higher c-coverage is not necessarily biologically meaningful since it may align many unrelated proteins together. Therefore, we also use GO terms to measure functional consistency of an alignment. GO terms describe roles of proteins in terms of their associated biological process (BP), molecular function (MF), and cellular component (CC). We exclude root GO terms from analysis, that is, GO terms on level higher than 5. We also exclude CC because proteins with matched CC are not usually considered functionally similar. Moreover, CC only annotates a small percentage of the proteins. The functional consistency measures specificity, average of functional similarity (AFS), mean normalized entropy (MNE), conserved orthologous interactions (COI), and sensitivity are based on the observation that functionally related proteins are more likely to have similar GO terms.
3.1. Alignment quality on real data
Topological quality: Supplementary Table S1 lists the topological evaluation of the competing methods. The first four multirows show the results for the clusters consisting of proteins belonging to c = 2, 3, 4, 5 species, respectively. In each multirow, the top and bottom rows show c-coverage and the number of proteins in the clusters, respectively. ConvexAlign has a larger c-coverage when c = 4, 5 than the other methods except SMETANA and NetCoffee. However, as we show later, many of the clusters generated by these two methods are not functionally conserved. The total coverage of BEAMS and IsoRank is better than the others because they produce many clusters composed of proteins from two or three species. These clusters cannot explain the data as well as clusters containing proteins from four or five species can. ConvexAlign has a better RCI than all other methods except SMETANA. These conserved interactions are very helpful in identifying the functional modules conserved among networks of different species.
Biological quality: Table 2 and Supplementary Table S2 provide the functional consistency measures of the competing methods. In the first four multirows, the top and middle rows show the number of consistent and annotated clusters, respectively, and the bottom row shows specificity. Regardless of c, ConvexAlign outperforms the other methods in terms of specificity and the number of consistent clusters. Although SMETANA, NetCoffee, and FUSE generate a larger number of clusters for c = 4, 5 than ConvexAlign, their clusters are not very functionally consistent. The fifth row shows that ConvexAlign has much higher specificity than the others when all the resulting clusters (c ≥ 2) are considered.
Note that for MNE, the smaller the better, while for the other measures, the larger the better.
CI, conserved interactions; COI, conserved orthologous interactions; MNE, mean normalized entropy.
These results suggest that ConvexAlign finds more functionally consistent clusters, not only by generating small clusters (i.e., c = 2, 3) but also more important large clusters (i.e., c = 4, 5). These clusters (especially when c = 4, 5) are very valuable because they may provide useful information about the orthology relationship among the proteins, identifying conserved subnetworks and predicting the function of unannotated proteins. In the sixth multirow, the bottom row shows the ratio of COI to the number of conserved interactions. ConvexAlign yields a COI/CI around 60% (i.e., 1.44 times larger than the second best ratio by BEAMS), indicating that ConvexAlign is able to identify conserved interactions between orthologous proteins. It also suggests that although SMETANA has the largest RCI, many of those conserved interactions are possibly formed by nonorthologous proteins. ConvexAlign also outperforms other methods in terms of MNE and sensitivity.
Table 3 shows the
We also apply ConvexAlign without using fedge (i.e., λ2 = 0) to show how edge preserving will improve the alignment accuracy. When λ2 = 0, the specificity is equal to 0.29 when c = 2 and is less than 0.50 when c = 3, 4, 5. Moreover, RCI = 0.01 and COI/CI = 0.16. These results indicate that preserving the edges may cause the nodes in dense subnetworks to align with each other. Thus, by applying fedge, ConvexAlign may align the similar modules together resulting in a more functionally meaningful alignment.
3.2. Alignment quality on synthetic data
Figure 1 shows the number of consistent clusters generated by different methods and their specificity on clusters composed of proteins in c = 2, 3, 4, 5, 6, 7, 8 species, respectively. In terms of the number of consistent clusters, ConvexAlign is slightly better than the second best method BEAMS regardless of c, but much better than the others. In terms of specificity, ConvexAlign has a much larger advantage over the other methods when c = 4, 5, 6, 7, 8. Fuse finds none or a few (1 or 2) consistent clusters when c = 3, 4, 5, 6. These results indicate that ConvexAlign aligns proteins in a functionally consistent way, without generating too many spurious clusters in which the proteins appear to be unrelated. Supplementary Figures S2 and S3 show that ConvexAlign outperforms all the other methods in terms of MNE, COI, and RCI.
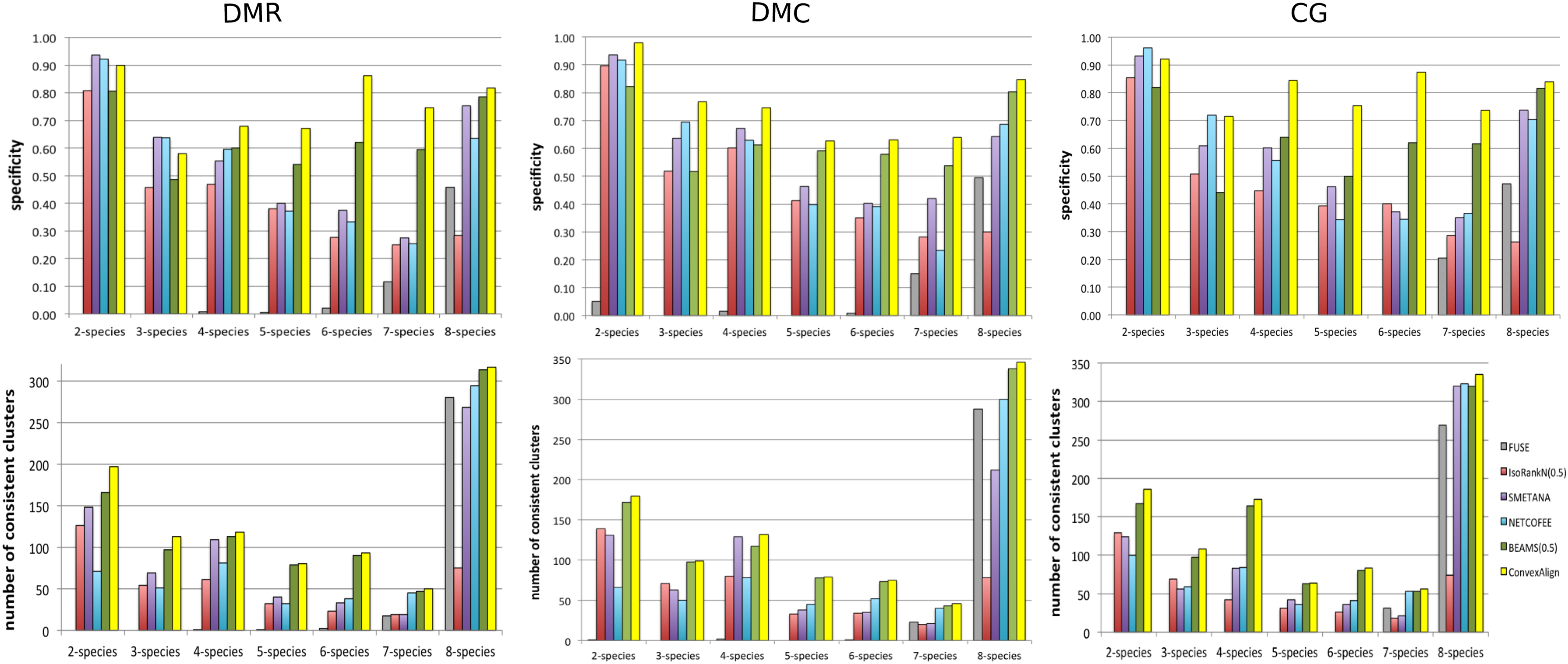
Specificity and the number of consistent clusters generated by the competing methods for different c on synthetic data. Only the best performance for IsoRankN and BEAMS is shown.
3.3. Finding conserved subnetworks
One of the applications of network alignment is to reveal subnetworks conserved across the species. These subnetworks are helpful for extracting biological information that cannot be inferred from sequence similarity alone. Figure 2 shows one conserved complex detected by ConvexAlign among the five species, but not appearing in the alignments generated by other methods. This complex is enriched for proteasome (with p-value< 10−7), which is essential for the degradation of most proteins, including misfolded or damaged proteins.
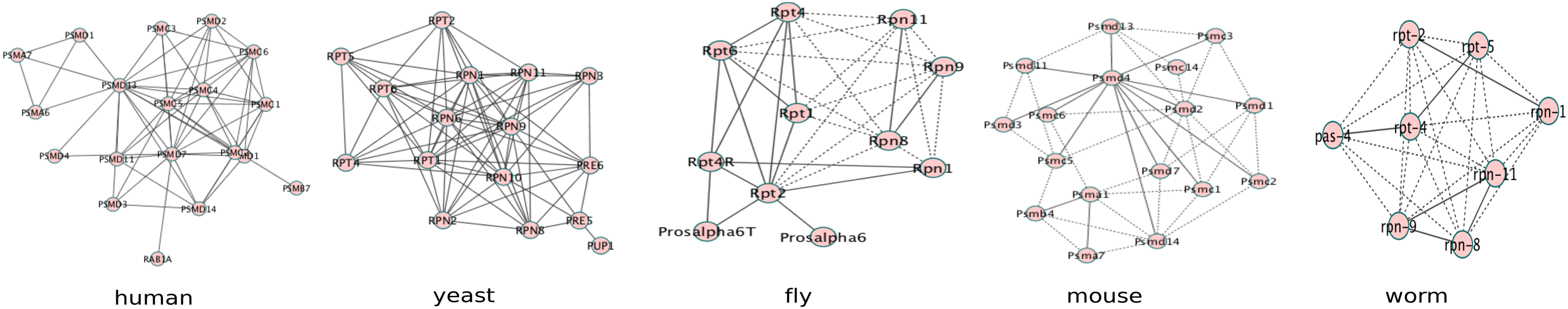
The ConvexAlign-detected proteasome complex in each input PPI network. PPI, protein–protein interaction.
In Figure 2, the interactions in IntAct are displayed in solid lines. For fly, mouse, and worm, some edges (shown by dotted lines) are missing in IntAct but present in the STRING database (Szklarczyk et al., 2011) with experimental evidence at the highest confidence. Note that our input networks consist of interactions only from IntAct but not STRING. This suggests that ConvexAlign is able to predict missing interactions. PANTHER (Mi et al., 2010) suggests that most of the aligned proteins are least divergent orthologs (Supplementary Table S3). There are some missing proteins from different species in some of the clusters. This is because either there are no orthologs in those species or there is no alignment for them. For example, cluster 5 has no proteins from worm and fly. PANTHER could not find any orthologous protein in those species either. Cluster 6 misses orthologous proteins in fly and yeast, which are aligned by ConvexAlign to proteins not in this proteasome complex. In addition, this proteasome complex has different number of nodes in different species, which implies that ConvexAlign is able to deal with inserted and deleted nodes.
Figure 3 shows another conserved subnetwork detected only by ConvexAlign that is related to DNA replication (with p-value<6−10). PANTHER suggests that the aligned proteins are orthologous and functionally related (Supplementary Table S4). It is worth mentioning that none of the abovementioned clusters appears in the alignment generated when λ2 = 0, indicating that edge preserving improves finding common functional/structural modules in the networks.
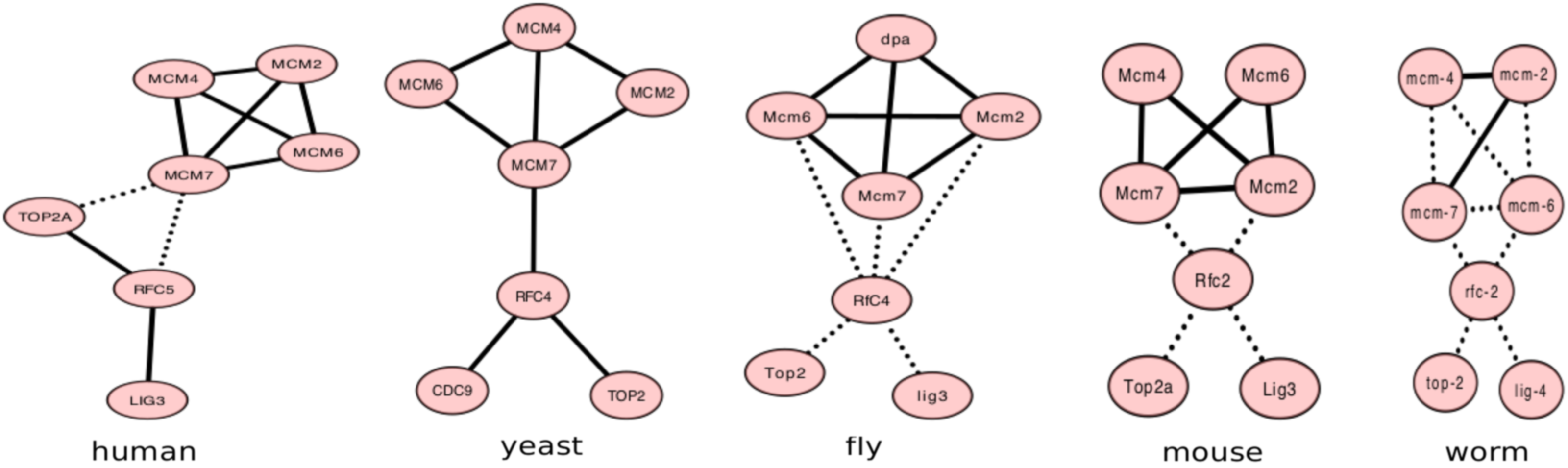
The ConvexAlign-detected DNA replication complex in each input PPI network.
3.4. Running time
ConvexAlign is computationally efficient compared to the other methods. Tested on the alignment of the networks of five species, it takes ConvexAlign, IsoRankN, BEMAS, FUSE, NetCoffee, and SMETANA 480, 1129, 900, 780, 15, and 37 minutes, respectively, to terminate. Although NetCofee has the smallest running time, it does not yield alignments with significant functional consistency.
4. Discussion
This article presents a new method, ConvexAlign, for global alignment of multiple PPI networks. ConvexAlign describes a new scoring function that integrates sequence and topological similarity between the matched proteins and interaction consistency. ConvexAlign uses a novel convex formulation to simultaneously align all the proteins in multiple input networks, resulting in better alignment quality. Such a formulation allows us to use an alternating direction method of multipliers (ADMM) method to find its optimal solution. We have tested ConvexAlign on real PPI networks, and the synthetic data evaluated the output alignments by different performance metrics and compared it to state-of-the-art methods. Experimental results show that on average ConvexAlign generates more functionally consistent clusters consisting of proteins from most of the input species. That is, ConvexAlign can explain a larger amount of data in a more functionally meaningful way. ConvexAlign can also find a few conserved and biologically important complexes that cannot be detected by the other alignment methods.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.